Download presentation
Presentation is loading. Please wait.

1
Regulatory Aspects of Clinical Trials in Malaysia
Centre for Investigational New Product National Pharmaceutical Control Bureau, MOH

2
Clinical Research and Compliance Section
OUTLINE Introduction Guidelines and Legal Requirements Application Process Audit and Inspection

3
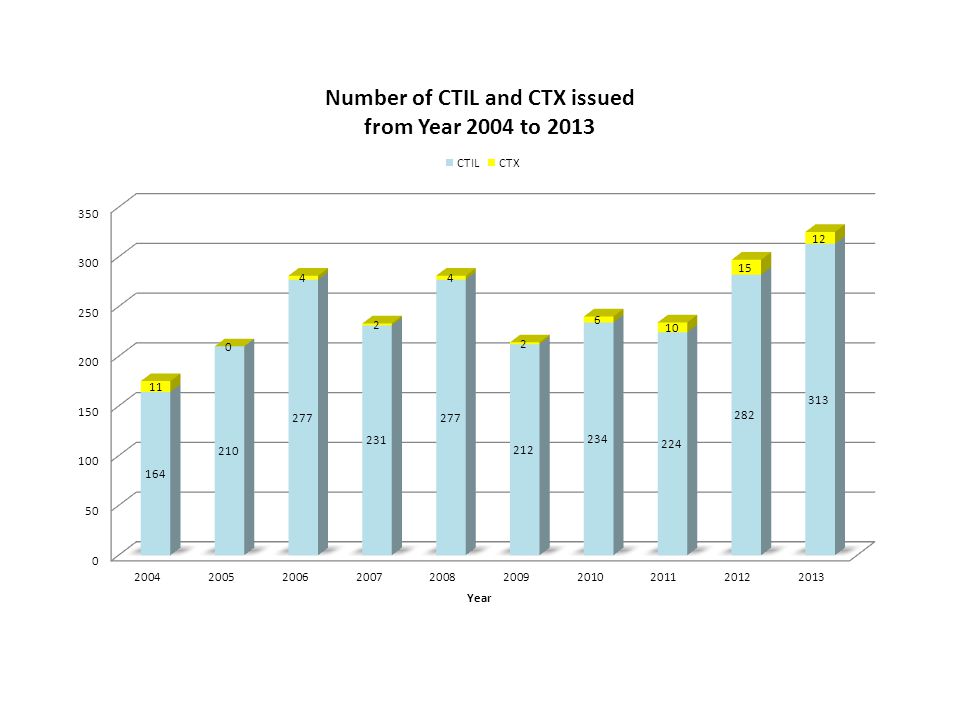
*Note These statistics are based on the number of Clinical Trial Import License and Clinical Trial Exemption applications received by National Pharmaceutical Control Bureau. Drug-related clinical trials for registered products which do not require clinical trial import license is not controlled by the Drug Control Authority.

4
*Note These statistics are based on the number of Clinical Trial Import License and Clinical Trial Exemption applications received by National Pharmaceutical Control Bureau. Drug-related clinical trials for registered products which do not require clinical trial import license is not controlled by the Drug Control Authority.

8
Guidelines and Legal Requirements
Malaysian Guidelines for Good Clinical Practice 3rd Edition (Updated Oct 2011, NPCB website) Guidelines for Application of CTIL and CTX in Malaysia 5th Edition (Updated June 2009, NPCB website) Guidelines for Good Clinical Practice Inspection (1st Edition Oct 2010)
 Guidelines for Application of CTIL and CTX in Malaysia 5th Edition (Updated June 2009, NPCB website) Guidelines for Good Clinical Practice Inspection (1st Edition Oct 2010)")
9
The Guidelines Should Be Read Together In Accordance To The Legal Requirements of ..
1. Control of Drugs and Cosmetics Regulations (CDCR) 1984 2. The Poison Regulations (Psychotropic Substances) 1989 3. Sale of Drugs Act 1952 where controlled medicines are involved
 The Poison Regulations (Psychotropic Substances) Sale of Drugs Act where controlled medicines are involved.")
10
In Malaysian Guidelines for GCP
1.55 Regulatory Authorities are bodies having the power to regulate, includes the authorities that review submitted clinical data that conduct inspection (Competent Authorities) 1.26 Drug Control Authority (DCA) An authority established for the purpose of regulating the Control of Drugs and Cosmetics Regulations, 1984
 1.26 Drug Control Authority (DCA) An authority established for the purpose of regulating the Control of Drugs and Cosmetics Regulations,")
11
Regulation 3(2) Control of Drugs and Cosmetics Regulations 1984 (Revised 2009) THE DRUG CONTROL AUTHORITY (DCA) The members are : the director-general of health (chairman) the director of pharmaceutical services (alternate chairman) the director of the National Pharmaceutical Control Bureau 8 other members appointed by the Minister of Health …..
 the director of pharmaceutical services (alternate chairman) the director of the National Pharmaceutical Control Bureau. 8 other members appointed by the Minister of Health …..")
12
Regulation 3(2) Control of Drugs and Cosmetics Regulations 1984 (Revised 2009) THE DRUG CONTROL AUTHORITY (DCA) …. The 8 other members appointed by the Minister of Health are : A consultant physician in the public service; A pharmacist in the public service 3 persons from any local universities with expertise in the pharmaceutical sciences 2 fully registered medical practitioners A veterinary practitioner in the public service

13
DCA’s Mission in Clinical Trials/ Research is Also Broad
Ensure Implementation of Good Clinical Practice (GCP) Standards GCP is an international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects GCP embraces trial objectives, trial design, study oversight, data collection and quality assurance, study analysis, as well as human subject protection in studies that support product applications
 Standards. GCP is an international ethical and scientific quality standard for designing, conducting, recording, and reporting trials that involve the participation of human subjects. GCP embraces trial objectives, trial design, study oversight, data collection and quality assurance, study analysis, as well as human subject protection in studies that support product applications.")
14
Objectives of Clinical Trial Assessment
Clinical Research and Compliance Section Objectives of Clinical Trial Assessment Protection of Clinical Trial Subjects Adequate disclosure of potential risks Trial has scientific merit CMC is Acceptable Regulations Regional & international guidelines Ethics Review Societal benefits from trial Data Integrity

15
CDCR 1984 (Revised 2009) Regulation 2
Definition of Product a drug in a dosage unit or otherwise, for use wholly or mainly by being administered to one or more human beings or animals for a medicinal purpose; or a drug to be used as an ingredient for a preparation for a medicinal purpose

16
SALES OF DRUGS ACT 1952 (Revised 1989)
Definition of Drug Includes any substance, product or article intended to be used or capable, or purported or claimed to be capable, of being used on humans or any animal, whether internally or externally, for medicinal purposes.

17
CDCR 1984 (Revised 2009) Regulation 7
Part III Registration and Licensing Regulation 7. Prohibition against manufacture, sale, supply, importation, possession and administration No person shall manufacture, sell, supply, import or possess or administer any product unless (a) the product is a registered product; and (b) the person holds the appropriate licence required and issued under these Regulations
 the product is a registered product; and. (b) the person holds the appropriate licence required. and issued under these Regulations.")
18
CDCR 1984 (Revised 2009) Notification of Cosmetics
Regulation 18A Prohibition to manufacture, sell, supply, import or process cosmetics No person sell manufacture, sell, supply, import or posses any cosmetic unless the cosmetic is a notified cosmetic. For the purpose of subregulation (1), “notified cosmetic” means a cosmetic which a notification note issued by Director of Pharmaceutical Services.
, notified cosmetic means a cosmetic which a notification note issued by Director of Pharmaceutical Services.")
19
CDCR 1984 (Revised 2009) Regulation 12
Regulation 12(1)(c): Clinical Trial Import Licence (CTIL) A Clinical trial import licence in Form 4 in the Schedule, authorising the licensee to import any product for purposes of clinical trials, notwithstanding that the product is not a registered product
(c): Clinical Trial Import Licence (CTIL) A Clinical trial import licence in Form 4 in the Schedule, authorising the licensee to import any product for purposes of clinical trials, notwithstanding that the product is not a registered product.")
20
CDCR 1984 (Revised 2009) Regulation 15 Exemptions
Regulation 15(5) : Clinical Trial Exemption (CTX) “Any person who wishes to manufacture any products solely for the purpose of producing samples for clinical trials, for registration or issuance of notification note under these Regulations may on application be exempted by the Director of Pharmaceutical Services from the provisions of regulation 7(1) or regulation 18A.”
 : Clinical Trial Exemption (CTX) Any person who wishes to manufacture any products solely for the purpose of producing samples for clinical trials, for registration or issuance of notification note under these Regulations may on application be exempted by the Director of Pharmaceutical Services from the provisions of regulation 7(1) or regulation 18A.")
21
CDCR 1984 (Revised 2009) Regulation 30
General Penalty (1) Any person who contravenes any of the provisions of these Regulations or any condition of any licence issued under these Regulations or any condition subject to which a product is registered under these Regulations commits an offence.
 Any person who contravenes any of the provisions of these Regulations or any condition of any licence issued under these Regulations or any condition subject to which a product is registered under these Regulations commits an offence.")
22
Section 12, SODA 1952 (Revised 1989)
INDIVIDUAL Section 12(2), SODA ’52 BODY CORPORATE First offence A fine not exceeding RM25,000 or to imprisonment for a term not exceeding three years or to both. A fine not exceeding RM50,000 Second or Subsequent offence A fine not exceeding RM50,000 or to imprisonment for a term not exceeding five years or to both. A fine not exceeding RM100,000
, SODA ’52. BODY CORPORATE. First offence. A fine not exceeding RM25,000 or to imprisonment for a term not exceeding three years or to both. A fine not exceeding RM50,000. Second or Subsequent offence. A fine not exceeding RM50,000 or to imprisonment for a term not exceeding five years or to both. A fine not exceeding RM100,000.")
23
Control of Drugs and Cosmetics Regulations 1984 (Revised 2009)
Regulation 29. Directions (1) The Director of Pharmaceutical Services may issue written directives or guidelines to any person or a group of persons as he thinks necessary for the better carrying out of the provisions of these Regulations and in particular relate to- (l) clinical trials or (2) Any person to contravenes any directives or guidelines issued by the Authority under subregulation (1) commits an offence.
 The Director of Pharmaceutical Services may issue written directives or guidelines to any person or a group of persons as he thinks necessary for the better carrying out of the provisions of these Regulations and in particular relate to- (l) clinical trials or (2) Any person to contravenes any directives or guidelines issued by the Authority under subregulation (1) commits an offence.")
24
Registration of Independent Ethics Committee with DCA
All Independent Ethics Committee approving drug related trial must be registered with the Drug Control Authority This directives was issued under Regulation 29, Control of Drugs and Cosmetics Regulations 1984 (Revised 2006)
")
25
Registration with National Medical Research Register (NMRR ) for all clinical trials that require CTIL/CTX A Directive had been issued effective from 1st January 2010, all Clinical Trial that require CTIL/CTX must register with NMRR, failure to register shall result in non-issuance of CTIL/CTX by DCA

26
Requirements to conduct BE studies for all generic products that contained controlled medicine and accreditation of the BE centres (1) A directive had been issued effective from 1st Jan 2012, all new generic products that apply for registration must have a BE study. A directive had been issued for those generic products that want to renew their registration must have a BE studies for renewal after 31st Dec 2012.

27
Requirements to conduct BE studies for all generic products that contained controlled medicine and accreditation of the BE centres (2) All BE centres that conduct BE studies must be accredited by NPCB effective from 1st Jan 2012 for both local and oversea BE centres.

28
Notification by sponsor/ BE centre to NPCB for all BE studies that do not required CTIL/CTX
A directive had been issued effective from 1st Jan 2012: For local pharmaceutical company, notification must be given to NPCB if they want to conduct the BE studies either locally (for generic product that do not required CTIL/CTX) / oversea. For foreign pharmaceutical company that conduct BE studies oversea must fulfill certain requirements before product registration.
 / oversea. For foreign pharmaceutical company that conduct BE studies oversea must fulfill certain requirements before product registration.")
29
Requirement on Drug-related Clinical Trial to register with NMRR
The previously issued directive has expanded as follows: All clinical trials involve drug has to be registered with NMRR. For the IEC/IRB provide ethical approval for clinical trials without registration with NMRR, will result its registration with DCA being suspended.

30
Guidelines were issued under Regulation 29, Control of Drugs and Cosmetics Regulations 1984 (Revised 2009) Guidelines for Application of CTIL and CTX in Malaysia 5th Edition (Updated June 2009, NPCB website) Guidelines for Good Clinical Practice Inspection (1st Edition Oct 2010)
 Guidelines for Good Clinical Practice Inspection (1st Edition Oct 2010)")
31
CTIL and CTX Application
Type of Application CTIL APPLICATION CTX APPLICATION Categories of Product Unregistered products when used or assembled (formulated or packaged) in a way different from the approved form for the purpose of Clinical Trial A traditional product with a marketing authorisation with indication for "traditionally used" when used for unapproved indication/ therapeutic claims for clinical trial purpose. Unregistered products to be imported Unregistered products to be manufactured locally Application form Borang BPFK 442.9 Borang BPFK 443.5 Fees RM 500 for each product Free Pharmacist Required? Licence A for Poisons /drugs (where applicable) NMRR Reg No. NMRR Registration No. License/ Permit Issuance Approval from DCA Approval from IEC/IRB
 in a way different from the approved form for the purpose of Clinical Trial. A traditional product with a marketing authorisation with indication for traditionally used when used for unapproved indication/ therapeutic claims for clinical trial purpose. Unregistered products to be imported. Unregistered products to be manufactured locally. Application form. Borang BPFK Borang BPFK Fees. RM 500 for each product. Free. Pharmacist Required Licence A for Poisons /drugs (where applicable) NMRR Reg No. NMRR Registration No. License/ Permit Issuance. Approval from DCA. Approval from IEC/IRB.")
32
CTIL/ CTX Requirements: (1) Who Can Apply?
Any Principal Investigator An authorised person from a LOCALLY REGISTERED PHARMACEUTICAL COMPANY (sponsor) / Clinical Research Organization with a permanent address in Malaysia *Note: Application for CTIL/ CTX containing a ‘poison/drug’ should be made by a LICENSE A HOLDER.
 / Clinical Research Organization with a permanent address in Malaysia. *Note: Application for CTIL/ CTX containing a ‘poison/drug’ should be made by a LICENSE A HOLDER.")
33
Note: The holder of CTIL/CTX need not necessarily conduct the clinical trial himself/herself The PI / Sponsor is allowed to submit parallel application to the DCA and IEC A CTIL will only be issued when both approval from DCA and IEC/IRB are obtained

34
CTIL/CTX Requirements: (2) Supporting Documents
Clinical Trial Protocol 1. Annex A Pharmaceutical Data 2. Annex B Investigator’s Brochure 3. Annex C

35
Requirements (1) Annex A- Clinical Trial Protocol
Name and dosage form of product Title and aim of the trial Description of the trial design Description of the subjects Treatment profile Operational aspects Adverse events Evaluation of results Approval by the principal investigator of the institution(s) where the clinical trial is to be done.
 where the clinical trial is to be done.")
36
Requirements (2) ANNEX B- QUALITY data of the investigational product
GMP statement from manufacturing / Certificate from Regulatory body Certificate of analysis Stability data (storage conditions) Manufacturing data & formulation Product labeling (coded & labeled: blinding)
 Manufacturing data & formulation. Product labeling (coded & labeled: blinding)")
37
Requirement 3: Annex C (Investigator’s Brochure)
CONTENT Safety Data of IP Efficacy Data of IP Non-Clinical Studies Pharmacology; PK/PD studies Toxicology Studies Marketing Experience, PSUR, product status Risks and ADR anticipated PK/PD studies in human In-house preliminary data Summaries of clinical trial conducted (Phase I, II, III) Published clinical data
 Published clinical data.")
38
Responsibility of the applicant
Responsible for the product and all information supplied for the CTIL/CTX application and updating the information If a service of CRO is used, a letter /authorization should be submitted to DCA Any person who knowingly supplies any false or misleading information in connection with his application for CTIL/CTX commits an offence under Control of Drugs and Cosmetics Reg.1984

39
Safety Decision Arising from Report Analysis/by Other Regulatory Authority
The DCA requires the sponsor to report within 48 hours of any significant safety issues which has arisen from an analysis of overseas reports or action with respect to safety which has been taken by another country’s regulatory agency. Sponsors should inform any Malaysian investigator(s) and, through the investigator, the IEC of this information. The DCA also requires that sponsors be able to provide promptly clinical details of any individual overseas adverse drug reaction reports if requested.
 and, through the investigator, the IEC of this information. The DCA also requires that sponsors be able to provide promptly clinical details of any individual overseas adverse drug reaction reports if requested.")
40
Clinical Trial Approval
A requirement in many countries Procedure varies Legislation vs non legislation

41
Factors Affecting Approval
The speed of approval depends on:- How complete is the information submitted? How fast sponsor/ PI respond to queries ? Enquiry should be answered within 30 working days. Failure with this requirement, CTIL/CTX will be rejected. Adherence to established procedures Ethical Approval

42
TIMELINE FOR APPROVAL IN MALAYSIA
EC, MOH 4 – 8 weeks Universities National Heart Institute 3 – 6 weeks DCA 30 working days* *Note: except for first in man trial, advanced therapy medicinal product (ATMP), Biotechnology product and Herbal products.
, Biotechnology product and Herbal products.")
43
CONDITIONS FOR CTIL/CTX IN THE GUIDELINES FOR APPLICATION OF CTIL/CTX IN MALAYSIA
CTIL Valid for 3 years (Regulation 12(5) of the CDCR 1984) Renewal of CTIL should be made within 3 months of the expiry date. Endorsement of CTIL/CTX-evidence of importation & delivery of the product to the investigator(s) Reporting of Suspected Unexpected Serious Adverse Reaction (SUSAR) Changes of Information
 of the CDCR 1984) Renewal of CTIL should be made within 3 months of the expiry date. Endorsement of CTIL/CTX-evidence of importation & delivery of the product to the investigator(s) Reporting of Suspected Unexpected Serious Adverse Reaction (SUSAR) Changes of Information.")
44
Discontinuation of trial with reasons. CTIL/CTX should be returned
End of Study Summary, Interim & Final Study Report Drug Accountability/Disposal Records/document : shipment, receipt System for retrieving and documentation System for the disposition of unused investigational products Archiving -responsibility of the investigator and the sponsor to archive safely all documents related to the trial

45
Audit & Inspection 5.19 of M’sian GCP Guidelines
By the local Regulatory Authority External Regulatory Authorities e.g: FDA,USA EMA,Europe

46
Audit & Inspection Audit Inspection What is the difference?

47
In the Malaysian Guidelines for GCP 1.7 What is an Audit?
A systematic and independent examination of trial related activities and documents to determine whether the evaluated trial related activities were conducted, and the data recorded, analyzed, and accurately reported according to the protocol, sponsor’s SOPS, GCP, the applicable regulatory requirement (s).
.")
48
In the Malaysian Guidelines for GCP 1.34 What is an inspection ?
The act by regulatory authority (ies) of conducting an official review of documents, facilities, records, and any other resources that are deemed by the authority (ies) to be related to the clinical trial and that may be located at the site of the trial, at the sponsor’s and / or Contract Research Organization’s (CRO’s) facilities, or at other establishments deemed appropriate by the regulatory authority (ies).
 of conducting an official review of documents, facilities, records, and any other resources that are deemed by the authority (ies) to be related to the clinical trial and that may be located at the site of the trial, at the sponsor’s and / or Contract Research Organization’s (CRO’s) facilities, or at other establishments deemed appropriate by the regulatory authority (ies).")
49
Audit & Inspection Audits = Sponsor function
Inspections= Regulatory function Generally, sponsor audits are conducted along similar lines to a regulatory inspection

50
Aims of Regulatory Inspections:
To determine the right, safety and well-being of a study subject has been protected. To determine whether the trial was conducted in accordance with applicable regulatory requirements, ethical standards and Malaysian Guidelines for Good Clinical Practice. To determine whether the data submitted in the dossier are credible and accurate. To assure the integrity of scientific testing and study conduct.

51
The USFDA has conducted 16 inspections of clinical studies in Malaysia
The USFDA has conducted 16 inspections of clinical studies in Malaysia. (*based on Clinical Trial in South East Asia (CTSE) 2012 Brief report on Thailand and Malaysia , presented at CTSE Conference and Yearly Update, 23rd November 2012 at Mumbai, India)
 2012 Brief report on Thailand and Malaysia , presented at CTSE Conference and Yearly Update, 23rd November 2012 at Mumbai, India)")
52
USFDA has conducted 16 clinical study inspections in Malaysia, no official action indicated in any of them

54
The highest number of findings in the inspections were related to protocol compliance and informed consent

55
A total of 6 large institutes in Malaysia were inspected by the USFDA, many of them multiple times

56
CLASSIFICATION OF INSPECTION FINDINGS/OBSERVATIONS
CRITICAL Conditions, practices or processes that adversely affect the rights, safety or well being of the subjects and/or the quality and integrity of data. Critical observations are considered totally unacceptable. Possible consequences: rejection of data and/or legal action and/or regulatory action required. Remark: Observations classified as critical may include a pattern of deviations classified as major, bad quality of the data and/or absence of source documents. Fraud belongs to this group.

57
MAJOR Conditions, practices or processes that might adversely affect the rights, safety or well-being of the subjects and/or the quality and integrity of data. Major observations are serious deficiencies and are direct violations of GCP principles. Possible consequences: rejection of data and/or regulatory action required. Remark: Observations classified as major, may include a pattern of deviations and/or numerous minor observations.

58
MINOR Conditions, practices or processes that would not be expected to adversely affect the rights, safety or well being of the subjects and/or the quality and integrity of data. Possible consequences: Observation classified as minor, indicate the need for improvement of conditions, practices and processes. Remark: Many minor observations might indicate a bad quality and the sum might be equal to a major finding with its consequences.

59
FDA Case Study #1: Impact of Inspection
Drug X in Long-Term Treatment of Condition Y Objective of the study to test the efficacy of drug X in outpatients when compare to placebo, as measured by the number of days until relapse Basis for site selection: site Eastern Europe showed a significant treatment response

60
FDA inspectional findings: there were in-patient hospitalizations for 24 subjects out of 35 subjects enrolled. DSI recommended to review division to reject data from this site.

61
Definition of Fraud Three general types of fraud: 1. ALTERED DATA
Generating biased data or changing data is legitimately obtained. 2. OMITTED DATA Not reporting data which has an impact on the study outcome E.g. Removing subjects from study population during analysis Not reporting or disguising adverse events 3. MANUFACTURED DATA Fabricating information or creating results without performing the work Filling in data in the CRF when work is not done Photocopying data and using it for multiple subjects Creating fictitious subjects

62
THANK YOU

Similar presentations






 is a review committee established to help protect the rights and welfare of human research subjects.>")



>")











