Download presentation
Presentation is loading. Please wait.

1
TRIGGER Trial: Transfusion in Gastrointestinal Bleeding
Investigator Meeting NHSBT – Birmingham August 29th 2012 11:00-16:30 Presented by Dr Vipul Jairath Research Fellow and Honorary SpR Gastroenterology NHSBT and Oxford University Hospitals

2
TRIGGER Investigator Meeting
Welcome Introductions Apologies Study Materials Site Folder Screening Log and CRFs Study Handbook

3
Overview of Policy Implementation, Screening and Consent Procedures
Vipul Jairath, Ana Mora, Sian Davies 11:30 – 12:15

4
Overview of trial recruitment processes
Participant admitted to hospital with suspected AUGIB Algorithm is followed for all patients except those where the clinician feels immediate transfusion is needed when the patient presents, regardless of or prior to obtaining the Hb result, due to severity of bleeding Follow the hospital’s allocated transfusion policy – Restrictive or Liberal Screen and Complete Screening Log Daily If participant declines consent for data use and follow-up, the algorithm is still followed Approach Eligible Patients to seek Consent Complete CRFs for Consenting Patients Collect data – discharge/death/D28 Telephone follow-up at Day 28

5
The Screening Log Each site should develop their own methods of case-ascertainment Log should be completed daily Monday – Friday Should record all new admissions with Acute Upper Gastrointestinal Bleeding (AUGIB) defined by: - Haematemesis: vomiting of blood, blood clots or witnessed coffee grounds - Melaena: passage of dark tarry stools either on clinical history witnessed by medical or nursing staff, or discovered on rectal examination
 defined by: - Haematemesis: vomiting of blood, blood clots or witnessed coffee grounds. - Melaena: passage of dark tarry stools either on clinical history witnessed by medical or nursing staff, or discovered on rectal examination.")
6
The Screening Log Screening Log (a): complete for ALL admission with AUGIB Screening Log (b): only complete for patients considered ‘Ineligible due to severity of bleeding’ See Screening Log forms (a) and (b) and Study Handbook pages 10 – 11 Send log through every Monday to CSU
: only complete for patients considered ‘Ineligible due to severity of bleeding’ See Screening Log forms (a) and (b) and Study Handbook pages 10 – 11. Send log through every Monday to CSU.")
7
Consent – for what? Randomisation – from the cluster
Intervention – from the cluster Data collection – from the participant Follow-up – from the participant

8
Consent – what is capacity?
Mental Capacity Act 2005, Section 2 ‘... a person lacks capacity if at the material time he is unable to make a decision for himself in relation to the matter because an impairment of, or disturbance in, the functioning of the mind or brain’ The following factors have to considered when assessing if someone has capacity to make a decision, section 3 (1): - whether they are able to understand the information - whether they are able to retain the information related to the decision to be made - whether they are able to use or weigh that information as part of the process of making the decision - whether they are able to communicate that decision – by any means
: - whether they are able to understand the information. - whether they are able to retain the information related to the decision to be made. - whether they are able to use or weigh that information as part of the process of making the decision. - whether they are able to communicate that decision – by any means.")
9
Consent procedures Spectrum of illness Anticipate that most patients will be able to provide consent for themselves Patients who may lack capacity include: - elderly patients with pre-existing cognitive impairment Patients who may have fluctuating capacity include: - elderly patients with acute confusional state - decompensated liver disease - alcohol withdrawal – especially Delerium Tremens

10
When and How to approach patients
Suggested timeframe to approach eligible patients/delegate: - on the same day for those who present in normal hours - the following day for those who present overnight - Monday for those who present over the weekend ... provided this is appropriate to their clinical condition Please use the suggested script as a guide which may help explain the transfusion policy – page 15 of Study Handbook Remember – you cannot complete any paperwork other than the anonymised screening log until you have written patient consent

11
Definitions - England Personal Consultee Nominated Consultee
- ‘... someone whom the person who lacks capacity would trust with important decisions about their welfare, but who is not acting in a professional or paid capacity, e.g you could consult a family member or close friend of the prospective trial participant, but not a paid carer or other professional such as social worker. Remuneration does not cover family members receiving some of the person’s pension or other benefits as a payment towards their share of the household expenses’ Nominated Consultee - ‘... Someone who is prepared to be consulted by the researcher, but has no connection with the research study, e.g this could be the consultant responsible for the patient’s care, provided they are not a named local PI, and should be of consultant (or equivalent) status. They must have received information about the research and a PIS is considered appropriate’
 status. They must have received information about the research and a PIS is considered appropriate’")
12
Definitions - Scotland
Relative/Welfare Attorney - ‘... Someone who has been appointed by a court to make decisions on behalf of those lacking capacity’

13
Consent Procedures – England (p.13 SH) CRF Form 1b
Does the patient have mental capacity in relation to the study? Yes Approach patient and use information sheet and consent form for participants with capacity No Does a personal consultee exist who can be approached in person? Yes No Approach nominated consultee. Use clinician agreement form Approach personal consultee. Use personal consultee information sheet and declaration form Does the patient regain mental capacity in relation to the study? Use declaration provided by personal consultee No Yes Use information sheet & consent form for participants regaining capacity and data preservation form

14
Consent Procedures – Scotland (p.14 SH) CRF Form 1b
Does the patient have mental capacity in relation to the study? Yes Approach patient and use information sheet and consent form for participants with capacity No Does a relative/welfare attorney exist who can be approached in person? Yes No Patient is not enrolled into study Approach relative/WA. Use relative/WA information sheet and declaration form Does the patient regain mental capacity in relation to the study? Use consent provided by relative/WA No Yes Use information sheet & consent form for participants regaining capacity and data preservation form

15
When is a Data Preservation form needed?
All patients who lacked capacity, and proxy consent (consultee/relative/welfare guardian) was obtained, must be approached once they regain capacity Use Data Preservation form (DPF) if: - a patient who regains capacity (and was enrolled on the basis of proxy consent) then declines to take part further in the study. Use of a Data Preservation Form will allow use of data up to that point, provided the patient agrees to this - a proxy initially provides consent and then decides to with draw this. Use of a Data Preservation Form will allow use of data up to that point, provided the proxy agrees to this
 was obtained, must be approached once they regain capacity. Use Data Preservation form (DPF) if: - a patient who regains capacity (and was enrolled on the basis of proxy consent) then declines to take part further in the study. Use of a Data Preservation Form will allow use of data up to that point, provided the patient agrees to this. - a proxy initially provides consent and then decides to with draw this. Use of a Data Preservation Form will allow use of data up to that point, provided the proxy agrees to this.")
16
Special Considerations - Scotland
Any participant with a pre-existing condition which would render them unable to provide consent would preclude them from participating in the study, e.g dementia Neither patients nor relatives/welfare guardians should be approached in this circumstance

17
Patients discharged prior to consent
Some centres are more pro-active at discharging lower risk patients from the ED Those will need to attempt to identify those patients and indicate this option on the weekly screening log In Scotland - post a cover letter and PIS/Consent Form plus stamped return envelope In England same approach applies but you will require R&D approval before you can implement this and these are pending at this time

18
Patients who die prior to consent
We anticipate this in very few circumstances Currently no approval process in place at this time and under REC review England – anticipate writing to relative >1 month after the event; this is under review and will not be in place for the start of the trial Scotland – under review

19
Trial Participant Checklist (p.8 SH)
Screening Log Consent – document in notes and 3 copies Insert algorithm into medical and nursing notes and adjacent to blood prescription chart Inform member of the clinical team of enrolment Inform Blood Bank if you have a flagging system Record preferred contact number(s) Post GP letter Start completing CRFs 1 – 10b, 15 – 17 Contact GP surgery to check survival status – D28 Administer telephone follow-up (Forms 11a – 13f) Complete End of Study form (Form 14)
 Post GP letter. Start completing CRFs 1 – 10b, 15 – 17. Contact GP surgery to check survival status – D28. Administer telephone follow-up (Forms 11a – 13f) Complete End of Study form (Form 14)")
21
Special considerations when consenting patients with AUGIB: A first hand nursing perspective
Sian Davies – RN 11:30 – 12:15

22
Characteristics of patients who may present with AUGIB
Older patients Patients with liver cirrhosis/alcoholic liver disease Patients with gastrointestinal malignancy (oesophageal/gastric/pancreatic) Patients taking Antiplatelets/NSAIDS
 Patients taking Antiplatelets/NSAIDS.")
23
Patients with liver cirrhosis
After a GI bleed, patient’s with cirrhosis can decompensate and develop encephalopathy. This often resolves over hours once treatment commenced In some situations encephalopathy is chronic, resulting in persistent fluctuating levels of confusion. Direct consent in this situation is often not achievable

24
Patients withdrawing from alcohol
Some patients (may or may not have cirrhosis) may be withdrawing from alcohol If withdrawal symptoms are adequately controlled, gaining consent should be straightforward If withdrawal symptoms are severe, e.g Delerium Tremens, consent would need to be initially sought from a personal consultee/relative/welfare guardian Day 3-5 of admission is often the most challenging. It should be possible to re-approach the patient after this time
 may be withdrawing from alcohol. If withdrawal symptoms are adequately controlled, gaining consent should be straightforward. If withdrawal symptoms are severe, e.g Delerium Tremens, consent would need to be initially sought from a personal consultee/relative/welfare guardian. Day 3-5 of admission is often the most challenging. It should be possible to re-approach the patient after this time.")
25
Patients with malignancy
Under these circumstances there may be no issues with consenting the patient from a capacity perspective Patients may be taking high doses of Opioid medication which could effect their ability to consent Caution around time of approach would be advised after discussion with medical/nursing team

26
Patients taking Antiplatelets/NSAIDS
Antiplatelets – likely to be older patient with co-morbidity. In this case they could suffer with an acute confusional state. Consent would need to be sought from a personal consultee/relative/welfare guardian until confusion resolves NSAIDS – combination of younger and older patients. Most should be capable of consenting

27
Review of Case Report Forms
Vipul Jairath & Ana Mora 12:15 – 13:15

28
Review of CRFs Tour through each CRF other than follow-up questionnaire (11a – 13f) and SAE forms (16a&b & 17) which will be reviewed later in the day Open CRFs and Completion Guidance pages 16 – 42 of Study Handbook All CRFs are the same apart from the front page, page headers and the consent form 1b
 and SAE forms (16a&b & 17) which will be reviewed later in the day. Open CRFs and Completion Guidance pages 16 – 42 of Study Handbook. All CRFs are the same apart from the front page, page headers and the consent form 1b.")
29
Forms 1a&b, 2, 3 & 4 – ‘Batch 1’ These should be submitted together as the first batch Form 1a – Eligibility Form 1b – Consent England and Scotland Form 2 – Presenting Signs and Symptoms Form 3 – First recorded Laboratory Data Form 4 – Pre-existing Co-morbidities and Medication

30
Forms 5a – 10b, 15 – ‘Batch 2’ These should be submitted together as the second batch Form 5a&b – Medication administered in Hospital Form 6a&b – Hb Measurements and RBC Transfusion Form 7 – Protocol Deviation Form Form 8a&b – Endoscopy Record Form 9 – Acute Transfusion Reactions Form 10a&b – Clinical Outcomes during Hospital Admission Form 15 – Additional Resource Use

31
Forms 11a – 14, 18 – ‘Batch 3’ These should be submitted together as the third batch Forms 11a – 13f – Follow-up Questionnaire Form 14 – End of Study Form Form 18 – Investigator Declaration Form

32
Transmittal of CRFs Order of transmittal - Forms 1a – 4 at trial entry
- Forms 5a – 10b, 15 at Hospital Discharge - Forms 11a – 14, 18 at end of study SAE transmittal forms - 16a, 16b & 17 as they occur

34
Lunch 13:15-13:45

35
Investigator responsibilities and
Safety reporting Alison Deary Clinical Operations Manager NHSBT 13:45-14:15

36
Investigator Responsibilities
These are divided into several areas: Qualifications and agreements Resources Responsibilities to the Participant Ethics and Governance Record Keeping Safety Reporting

37
Qualifications and Agreements
GCP states that the investigator should be qualified by education, training and experience to assume responsibility for the proper conduct of the trial.. CV/GCP training certificate Familiar with the protocol and the appropriate use of the investigational product Delegation of trial-related duties

38
Qualifications and Agreements
Should be aware of, and comply with GCP and applicable regulatory requirements Should permit monitoring and audit by the sponsor and inspection by regulatory authority Should maintain a list of appropriately qualified persons to whom trial related duties are delegated

39
Resources Be able to demonstrate the potential to recruit the required number of suitable participants within the agreed recruitment period Have sufficient time Have adequate number of qualified staff and adequate facilities Ensure all staff are adequately informed

40
Responsibilities to the Participant
Medical care of the participants – make all trial related medical decisions Ensure adequate care for adverse events Make a reasonable effort to ascertain reasons for withdrawal from the trial Ensure participant confidentiality respected at all times Ensure participant’s medical records reflects their participation Inform GP

41
Ethics and Governance Ensure have ethical and trust approval to conduct the study Conduct study according to current version of the protocol Document and explain any deviation from the protocol Not to implement any deviation from study without permission from sponsor (and REC) unless immediate safety concern
 unless immediate safety concern.")
42
Ethics and Governance Obtain informed consent
Ensure using current version of information and consent forms Until consent obtained, no trial-related activity may commence Ensure a copy of information and consent form are provided to participant/representative Follow randomised trial treatment policy

43
Record keeping Obtain informed consent and document this
Ensure data collected are consistent with source documents Collect, record and report all study data accurately , completely, legibly and in a timely manner To sign the completed CRFs To ensure all trial documentation is maintained appropriately To ensure essential documents are archived To ensure annual reports are made to the R&D department

44
Safety Reporting

45
Adverse Event Definitions
An adverse event is defined as “Any untoward occurrence in a subject receiving treatment according to the protocol, including occurrences which are not necessarily caused by or related to administration of the research procedures”

46
Serious Adverse Events (SAE)
Defined as any adverse event that Results in death Is life-threatening Results in hospitalisation or prolongation of existing hospitalisation Results in persistent disability or incapacity Do not confuse serious with severe

47
Expected or Unexpected?
Section 9 of the protocol contains a list of “expected” SAEs Only unexpected SAEs – i.e. not on the list of “expected” SAEs - are subject to expedited notification to the CSU An unexpected related SAE is one that is judged to be possibly, probably or definitely related to the transfusion policy

48
Definitions of Imputability
Relationship Description Possible There is some evidence to suggest a causal relationship, however the influence of other factors may have contributed to the event Probable The evidence is clearly in favour of attributing the SAE to the transfusion policy Definite There is conclusive evidence beyond reasonable doubt attributing the SAE to the transfusion policy e.g. Because the event occurs within a reasonable timeframe after the transfusion or with-holding of the transfusion e.g. patients clinical condition or concomitant treatments

49
Expected SAEs which are clinical outcome measures
Death Further bleeding Need for surgery or radiological intervention to control bleeding Any element of the composite endpoint of thromboembolic and ischaemic events (MI, stroke, PE, DVT, acute kidney injury) Acute transfusion reaction
 Acute transfusion reaction.")
50
Other expected SAEs Multi-organ failure Congestive Cardiac Failure
Respiratory complications Transient ischaemic attack Decompensated liver disease Need for paracentesis to drain ascites Gut infarction

51
Other expected SAEs Rise in serum Troponin I or E
GI perforation or obstruction Post-operative complication Need for repeat endoscopy for any reason Non-acute transfusion reaction

52
Notification within one working day
Any unexpected SAE Death Further bleeding Any element of the composite endpoint of thromboembolic and ischaemic events (MI, stoke, PE, DVT, acute kidney injury)
")
53
Reporting to IDMC When received at the CSU, the reports of the events listed will be sent to the IDMC and the CI Be prepared to answer any queries in a timely manner The IDMC will know which treatment regimen the site has been randomised to

54
When to collect SAEs Collect on all participants from enrolment to Study Day 28 Include any that occur following discharge if before Day 28, in particular infection, further bleeding and thromboembolic/ischaemic events

55
Responsibilities -PI Ensure notification of all SAEs in the appropriate timeframe SAE forms must be completed by the PI, or, if a designate, counter-signed by the PI Initial report must contain at least patient identifier, name of event, date of occurrence and causality Further more detailed information can be provided later

56
Responsibilities - Sponsor
The CI will review all reported SAEs and Clinical Outcomes. Any disagreement with regard to imputability will be recorded but the PI opinion cannot be over-ruled. Unexpected related SAEs will be reported to REC within 15 days

57
Form Completion Review of Forms 16a&b, 17 SAE transmittal

59
Case-Study 1 68 year old female admitted with UGIB and found to have a large ulcer at endoscopy which is treated. Has further bleeding 72 hours later and a repeat endoscopy and therapy. Discharged home 5 days later uneventfully. How many SAEs in this scenario? Are they expected or unexpected? Within what timeframe should they be notified to the CSU?

60
Case-Study 1 Answer 68 year old female admitted with UGIB and found to have a large ulcer at endoscopy which is treated. Has further bleeding 72 hours later and a repeat endoscopy and therapy. Discharged home 5 days later uneventfully. How many SAEs in this scenario? - One Are they expected or unexpected? Expected Within what timeframe should they be notified to the CSU? Within 1 working day

61
Case-Study 2 45 year old female with alcoholic liver disease admitted with a variceal bleed. Varices successfully banded at endoscopy but she was combative during the procedure and it was prolonged. She then developed an aspiration pneumonia requiring antibiotics. She was discharged without further complications How many SAE’s are there in this scenario? Are they expected or unexpected? Within what timeframe should they be notified to the CSU?

62
Case-Study 2 Answer 45 year old female with alcoholic liver disease admitted with a variceal bleed. Varices successfully banded at endoscopy but she was combative during the procedure and it was prolonged. She then developed an aspiration pneumonia requiring antibiotics. She was discharged without further complications How many SAE’s are there in this scenario? One Are they expected or unexpected? Expected Within what timeframe should they be notified to the CSU? Within one week

63
Case-Study 3 54 year old male has a successfully treated duodenal ulcer at endoscopy. He has further bleeding and then goes straight to radiological embolisation. He has further bleeding and then has surgery. He is due for discharge 10 days later but develops a wound infection and is kept in for a further 4 days for antibiotics. How many SAE’s are there in this scenario? Are they expected or unexpected? Within what timeframe should they be notified to the CSU?

64
Case-Study 3 Answer 54 year old male has a successfully treated duodenal ulcer at endoscopy. He has further bleeding and then goes straight to radiological embolisation. He has further bleeding and then has surgery. He is due for discharge 10 days later but develops a wound infection and is kept in for a further 4 days for antibiotics. How many SAE’s are there in this scenario? Four Are they expected or unexpected? All expected Within what timeframe should they be notified to the CSU? Further bleeding within one working day; Embolisation/surgery/wound infection within one week

65
Case-Study 4 78 year old man previous heart attacks and “mini-strokes” taking life-long aspirin and dypiridamole. Found to have a gastric cancer at endoscopy. Anti-platelets are withheld and he develops a stroke with a dense right hemi-paresis. How many SAE’s are there in this scenario? Are they expected or unexpected? Within what timeframe should they be notified to the CSU?

66
Case-Study 4 Answer 78 year old man previous heart attacks and “mini-strokes” taking life-long aspirin and dypiridamole. Found to have a gastric cancer at endoscopy. Anti-platelets are withheld and he develops a stroke with a dense right hemi-paresis. How many SAE’s are there in this scenario? One Are they expected or unexpected? Expected Within what timeframe should they be notified to the CSU? Within one working day

67
Case-Study 5 A 32 year old alcoholic went on a binge and was vomiting for 24 hours. He was found to have a Mallory-Weiss tear at endoscopy. His alcohol withdrawal symptoms were severe and he fell on the ward whilst mobilising to the toilet, banged his head and required 4 stitches. How many SAE’s are there in this scenario? Are they expected or unexpected? Within what timeframe should they be notified to the CSU?

68
Case-Study 5 Answer A 32 year old alcoholic went on a binge and was vomiting for 24 hours. He was found to have a Mallory-Weiss tear et endoscopy. His alcohol withdrawal symptoms were severe and he fell on the ward whilst mobilising to the toilet, banged his head and required 4 stitches. How many SAE’s are there in this scenario? None! This is an adverse event, not a serious adverse event. Adverse events do not need reporting.

69
Health Economics Research Centre-University of Oxford
Health Economics Evaluations: What are they and what will we do in TRIGGER Elizabeth Stokes Researcher Health Economics Research Centre-University of Oxford 14:15-14:40

70
Overview of session Introduction to health economic evaluation
Rationale for it What it is How it is conducted and results interpreted Health economics in TRIGGER Data collection forms in TRIGGER Introduce economic evaluation and why it is important 70

71
What is health economic evaluation?
Premise: scarce (health care) resources Aim: to maximise health gain with the resources available Method: compare costs and outcomes of two or more interventions Definition: “The comparative analysis of alternative courses of action in terms of both their costs and their consequences” (Drummond et al. 2005) Explicit way of making choices Economics looks at what mechanisms we can use to allocate scarce resources. Sometimes known as the gloomy or dismal science as the idea that we can’t have everything that we want is at the basis of all our work. Resources are scarce, and we have to make choices about how to allocate these scarce resources. Resources – basic inputs to production – land, labour, capital. Scarcity – not enough resources to satisfy all demands/wants – 2 sides: infinite nature of human wants, finite nature of resources. NHS has a finite budget, and what we spend on 1 thing, we will not have to spend on something else. Methods - Costs and effectiveness of new interventions must be compared with existing interventions. Comparative. So economic evaluation is about costs and effects, inputs and outputs Explicit - it allows us to quantify the change in costs and the change in effects as we move from one treatment or intervention to another, and it provides a means of identifying which is the most cost-effective treatment or intervention, in other words the one which represents value for money. 71
 resources. Aim: to maximise health gain with the resources available. Method: compare costs and outcomes of two or more interventions. Definition: The comparative analysis of alternative courses of action in terms of both their costs and their consequences (Drummond et al. 2005) Explicit way of making choices. Economics looks at what mechanisms we can use to allocate scarce resources. Sometimes known as the gloomy or dismal science as the idea that we can’t have everything that we want is at the basis of all our work. Resources are scarce, and we have to make choices about how to allocate these scarce resources. Resources – basic inputs to production – land, labour, capital. Scarcity – not enough resources to satisfy all demands/wants – 2 sides: infinite nature of human wants, finite nature of resources. NHS has a finite budget, and what we spend on 1 thing, we will not have to spend on something else. Methods - Costs and effectiveness of new interventions must be compared with existing interventions. Comparative. So economic evaluation is about costs and effects, inputs and outputs. Explicit - it allows us to quantify the change in costs and the change in effects as we move from one treatment or intervention to another, and it provides a means of identifying which is the most cost-effective treatment or intervention, in other words the one which represents value for money. 71.")
72
Types of health economic evaluation
Costs Outcomes Cost-effectiveness analysis Monetary units Natural units e.g. cases detected, life years gained, episode-free day Cost-utility analysis Quality Adjusted Life Years (QALYs) Cost-benefit analysis All three forms of evaluation are the same in that they measure costs in monetary units, it being on the outcome side that they differ. CEA: outcomes or effects are measured in natural units, specific to what is being evaluated. E.g. studies comparing different diagnostic strategies for DVT have used cases detected as their outcome measure, studies comparing various treatments for prolonging life after renal failure have used life years gained, and studies comparing treatments for asthma have used episode-free days. CEA is perhaps most useful at a local level when an individual decision maker working with a budget is considering a range of treatments or interventions within a particular specialty or for a particular condition. It is not so useful for assessing the cost-effectiveness of interventions across specialties or conditions, because of the lack of a common outcome measure, for example one couldn’t directly compare an intervention where outcomes are measured using cases detected, with one where outcomes were measured as symptom free days. In this case, we need some generic or common measure of outcome, and we get this with CUA, where outcomes are measured using QALYs. The QALY is a composite measure of health outcome combining two things, firstly the impact that an intervention has on quantity of life or years of survival and secondly the impact it has on quality of life experienced through out those years of survival. The rationale behind the QALY is we give health care treatments and interventions either to improve patients’ survival prospects, to improve their HRQoL (so to make people feel better), or both, and therefore in theory the QALY can be used to capture the impact of any health care intervention. It is a generic measure of health outcome and this makes it attractive in that it can be used to measure and compare the effectiveness and cost-effectiveness of health care programmes across different specialties, from say, blood transfusion to hip replacement to cardiac surgery to cancer screening. It is because of these advantages, and the recent drive towards the need to demonstrate the cost-effectiveness of new health care interventions against those already funded, that cost-utility analysis is the most frequently conducted type of economic evaluation, and is the approach that we shall take in TRIGGER. I’ll talk more about QALYs on the next slide. Finally we have cost-benefit analysis, in which both costs and outcomes are valued in monetary units. The idea behind CBA is that it goes one step further than CUA, in that because the outcome is not specific to health, this form of analysis could be used to compare the value for money not just of competing health care interventions, but also of programmes or policies across other sectors of the economy for example in education, transport and so on. In theory therefore it could enable resources to be allocated efficiently across all sectors of the economy. In practice however, CBA is rarely conducted because of the difficulties that arise when attempting to place monetary values on health outcomes. For this reason, we shall set CBA aside, and say that what follows refers to CEA and CUA only. 72
 Cost-benefit analysis. All three forms of evaluation are the same in that they measure costs in monetary units, it being on the outcome side that they differ. CEA: outcomes or effects are measured in natural units, specific to what is being evaluated. E.g. studies comparing different diagnostic strategies for DVT have used cases detected as their outcome measure, studies comparing various treatments for prolonging life after renal failure have used life years gained, and studies comparing treatments for asthma have used episode-free days. CEA is perhaps most useful at a local level when an individual decision maker working with a budget is considering a range of treatments or interventions within a particular specialty or for a particular condition. It is not so useful for assessing the cost-effectiveness of interventions across specialties or conditions, because of the lack of a common outcome measure, for example one couldn’t directly compare an intervention where outcomes are measured using cases detected, with one where outcomes were measured as symptom free days. In this case, we need some generic or common measure of outcome, and we get this with CUA, where outcomes are measured using QALYs. The QALY is a composite measure of health outcome combining two things, firstly the impact that an intervention has on quantity of life or years of survival and secondly the impact it has on quality of life experienced through out those years of survival. The rationale behind the QALY is we give health care treatments and interventions either to improve patients’ survival prospects, to improve their HRQoL (so to make people feel better), or both, and therefore in theory the QALY can be used to capture the impact of any health care intervention. It is a generic measure of health outcome and this makes it attractive in that it can be used to measure and compare the effectiveness and cost-effectiveness of health care programmes across different specialties, from say, blood transfusion to hip replacement to cardiac surgery to cancer screening. It is because of these advantages, and the recent drive towards the need to demonstrate the cost-effectiveness of new health care interventions against those already funded, that cost-utility analysis is the most frequently conducted type of economic evaluation, and is the approach that we shall take in TRIGGER. I’ll talk more about QALYs on the next slide. Finally we have cost-benefit analysis, in which both costs and outcomes are valued in monetary units. The idea behind CBA is that it goes one step further than CUA, in that because the outcome is not specific to health, this form of analysis could be used to compare the value for money not just of competing health care interventions, but also of programmes or policies across other sectors of the economy for example in education, transport and so on. In theory therefore it could enable resources to be allocated efficiently across all sectors of the economy. In practice however, CBA is rarely conducted because of the difficulties that arise when attempting to place monetary values on health outcomes. For this reason, we shall set CBA aside, and say that what follows refers to CEA and CUA only. 72.")
73
Quality Adjusted Life Years (QALYs)
Composite endpoint combining: Quantity of life (survival) Health related quality of life (HRQoL) measured on a 0 (dead) to 1 (full health) scale Health related quality of life 1 Survival time (years) 2 0.6 0.4 0.3 0.8 Patient 1 = = 1.4 QALYs Patient 2 = = 0.55 QALYs QALY difference = 0.85 QALYs A bit more on QALYs. QALYS are increasingly used as the measure of outcome in CEA and are a composite measure of health outcome which combine 2 different things: firstly we look at the quantity of life - survival or years of life lived by individuals, and then secondly, we adjust for the level of HRQoL at which those years of life are lived, and we measure HRQoL on a scale from 0 to 1 where 0 is considered equivalent to death and 1 to perfect health. Using this graph here, I would like to illustrate to you what we call a QALY profile and how we go about calculating QALYs. On the vertical axis we are measuring HRQoL on our 0 to 1 scale, and on the horizontal axis, survival time in years. Lets assume we have a patient who we know survives for 2 years following some new intervention or treatment. We have measured the patient’s QoL and know that in the first year he is at a level of 0.8 whereas in the second year his QoL falls to The QALYs experienced by this patient are estimated simply by taking the area under the curve, so we have 0.8 * 1 = 0.8 QALYs in the first year, and 0.6*1, 0.6 QALYs in the second year, giving a total of 1.4 QALYs experienced over the two years survival period. Suppose now we have a second patient who received the comparator intervention or treatment. This patient survives for only 1.5 years, and has a much lower quality of life throughout, accruing just 0.55 QALYs, 0.4 QALYs in year 1, and then 0.15 QALYs thereafter. The QALY difference is therefore 0.85, and this is the area between the two QALY profile curves, and so takes into account the differences in quality of life and length of life. 73
 Health related quality of life (HRQoL) measured on a 0 (dead) to 1 (full health) scale. Health related quality of life. 1. Survival time (years) Patient 1 = = 1.4 QALYs. Patient 2 = = 0.55 QALYs. QALY difference = 0.85 QALYs. A bit more on QALYs. QALYS are increasingly used as the measure of outcome in CEA and are a composite measure of health outcome which combine 2 different things: firstly we look at the quantity of life - survival or years of life lived by individuals, and then secondly, we adjust for the level of HRQoL at which those years of life are lived, and we measure HRQoL on a scale from 0 to 1 where 0 is considered equivalent to death and 1 to perfect health. Using this graph here, I would like to illustrate to you what we call a QALY profile and how we go about calculating QALYs. On the vertical axis we are measuring HRQoL on our 0 to 1 scale, and on the horizontal axis, survival time in years. Lets assume we have a patient who we know survives for 2 years following some new intervention or treatment. We have measured the patient’s QoL and know that in the first year he is at a level of 0.8 whereas in the second year his QoL falls to 0.6. The QALYs experienced by this patient are estimated simply by taking the area under the curve, so we have 0.8 * 1 = 0.8 QALYs in the first year, and 0.6*1, 0.6 QALYs in the second year, giving a total of 1.4 QALYs experienced over the two years survival period. Suppose now we have a second patient who received the comparator intervention or treatment. This patient survives for only 1.5 years, and has a much lower quality of life throughout, accruing just 0.55 QALYs, 0.4 QALYs in year 1, and then 0.15 QALYs thereafter. The QALY difference is therefore 0.85, and this is the area between the two QALY profile curves, and so takes into account the differences in quality of life and length of life. 73.")
74
Cost-effectiveness framework
Intervention 1 Intervention 2 Cost 1 Cost 2 Effectiveness 1 Effectiveness 2 As I have mentioned previously, the approach to determining whether an intervention is cost-effective when carrying out an economic evaluation necessitates the measurement of costs and effects on two or more competing interventions. Intervention 1 is normally some new treatment or policy and intervention 2 is what we call the comparator intervention, it is the treatment or policy that we wish to compare our new intervention against. In most evaluations, the comparator intervention will be that which is being already carried out in routine practice. The key thing to emphasise is that we are interested in the differences between these two interventions, so what is the difference in cost and the difference in outcomes if we were to replace intervention 2, with intervention 1? In certain situations we also often relate the difference in costs to the difference in effects using something called the incremental cost-effectiveness ratio which is estimated by dividing the difference in costs by the difference in effects. To give you a published example……. Incremental cost-effectiveness ratio (ICER) = Cost 1 - Cost 2 Effect 1 – Effect 2 74
 = Cost 1 - Cost 2. Effect 1 – Effect")
75
Incremental Costs and Outcomes: Example – chronic low back pain
Surgery £7,830 1.004 QALYs Rehabilitation £4,526 0.936 QALYs £7,830 - £4,526 These are the cost and effect results from a study comparing surgical stabilisation of the spine with rehabilitation for patients with chronic low back pain. The study was a RCT and patients were followed up for 2 years; data was collected as part of the trial which would enable the costs and effects of each intervention to be estimated. The cost in the surgery arm was on average £7,830 per patient and surgery was associated with QALYs over the two years. Rehabilitation cost on average £4526 per patient and was associated with QALYs. Surgery therefore was on average more costly than rehabilitation but also more effective – a common situation which often prevails in cost-effectiveness analyses. Concluding that the intervention costs around £3,300 more and generates an additional QALYs is perhaps not so useful and so we kind of standardise the results using the incremental cost-effectiveness ratio. By dividing the difference in costs by the difference in effects, we can determine what the additional costs would be to generate 1 whole additional unit of outcome. In this case, close to £50,000 per additional QALY. As you will see in just a moment, it is this ICER which is key when determining whether an intervention is cost-effective. = £48,588 per QALY 1.004 QALY QALY Rivero-Arias et al. BMJ 2005; 330(7502): 1239 75
:")
76
Health Economics in TRIGGER
Feasibility trial Feasibility of gathering data required in a cost-effectiveness analysis to inform data collection for the phase III trial Pilot resource use data collection forms Assess feasibility of collecting inpatient data from routine hospital information systems, post-discharge data from patients by telephone, and HRQoL data using the EQ-5D Feasibility of using these data within a cost-effectiveness model to identify parameters with the potential to be key drivers of cost-effectiveness These will require detailed measurement in phase III trial Feasibility of using EQ-5D in this patient group – good response rate? Responses intuitive and complete? Feasibility of collecting data to allow us to estimate and compare the costs and effects of a restrictive versus a liberal transfusion policy in AUGIB patients. On the cost side, we are interested in what is the net cost impact of moving from a liberal to a restrictive transfusion policy. It is challenging a priori to determine what this might be. On the outcome side, we are interested in the net impact of a restrictive transfusion policy upon both length and quality of life. In particular we would hypothesise that patients for whom death is averted will likely survive into the future and continue to enjoy additional years of life that might otherwise have been lost. Taking into account the quality of life at which years of life are lived is also of interest. 76

77
Costs I Perspective of National Health Service, patients / their carers, and employers Data will be collected from each trial patient on inpatient health care resource use, including: RBCs and other blood products transfused Endoscopies Time spent in ICU, HDU, general wards Adverse events Discharge / transfer to another hospital / facility Data collection integrated into the CRFs Focussing in on the costing side of the study, the perspective we have decided to adopt is that of the National Health Service, patients, and employers. When we talk about perspective or viewpoint, we are basically talking about whose costs we are going to include and measure. In the past, the majority of cost-effectiveness studies adopted quite a narrow perspective and focussed only on direct health care costs. Nowadays though, economists are encouraged to include a wider range of costs especially if there is reason to believe they might vary between the interventions being evaluated. Inpatient resources ...., hopefully all of the main in-hospital cost drivers. 77

78
Costs II Primary and secondary health care resource use post discharge will be captured via a telephone contact with patients at 28 days Also in this questionnaire patients will be asked about any unpaid care received from relatives or friends and any costs they have incurred and about return to paid employment Unit costs from a variety of sources will be used to cost this resource use data. Additionally, to capture health care costs after hospital discharge, we shall use a telephone contact with the patient at day 28 to ask about their contacts with the primary and secondary health care sectors since discharge. Ask about hospital readmissions, outpatient visits, community care, GP visits Unpaid informal care / employment – determine if these are important to capture in the phase III trial Finally, once measured, we shall cost all of this resource use data using unit costs from a variety of sources including NHS Reference Costs, Personal and Social Services Research Unit. 78

79
Effects I Measured using Quality Adjusted Life Years (QALYs)
EuroQol EQ-5D questionnaire used to classify a person’s health state by telephone at day 28 EQ-5D asks respondents whether they have no problems, some problems or extreme problems with Responses are converted into a single index value on the scale 0 to 1, dead to perfect health, using an accompanying scoring tariff - Mobility - Self-care - Usual activities - Pain / discomfort - Anxiety / depression The effectiveness or outcomes associated with a restrictive versus a liberal transfusion policy in AUGIB patients will be measured using QALYs. We have selected the QALY as the outcome measure for a number of reasons. Firstly, the potential exists for the use of a restrictive transfusion policy in AUGIB patients to both reduce patient mortality and possibly have a positive impact upon HRQoL, effects which would both be captured by the QALY. Secondly, the QALY is a generic measure of health outcome and therefore it becomes possible to assess the value for money offered by health care interventions across different specialties, from say, blood transfusion to hip replacement to cardiac surgery to cancer screening. It also explains why in England and Wales, when assessing the value for money of new interventions and making recommendations on adoption to the NHS, NICE specifically requires that for cost-effectiveness analyses, the value of health effects should be expressed in terms of QALYs. HRQoL captured by the EQ-5D. For patients still in hospital at day 28, the EQ-5D will be completed in hospital. 79

80
Effects II Survival data available for each trial patient to 28 days
HRQoL assessed at 28 days using the EQ-5D questionnaire No baseline values due to unplanned nature of the condition Quality and quantity of life combined and used as inputs into decision model To facilitate the calculations of QALYs, we shall collect survival data for each trial patient at 28 days, and assess patient HRQoL at 28 days using the EuroQol EQ-5D questionnaire. No baseline HRQoL data, so we will likely have to make some assumption about levels of QoL at this time and test these assumptions in some kind of sensitivity analysis. Final step is to generate a QALY profile for each patient to 28 days and estimate the number of QALYs accrued by each patient. 80

81
Key drivers of cost-effectiveness
Trial data will allow exploration of mean costs and QALYs per patient for each trial arm to 28 days Feasibility trial, so the time horizon is short Model acute upper GI bleeding (AUGIB) patient pathway & associated costs and outcomes beyond 28 days to identify parameters with the potential to be key drivers of cost-effectiveness Use trial data, and other sources such as audit of AUGIB The same model will be used to model costs/ effects beyond 28 days to identify key cost drivers as part of the feasibility trial, as will be used to extrapolate costs and effects beyond the end of the trial time horizon in the phase III trial. But, models being used in different ways. Feasibility – lots of sensitivity analysis as lot of uncertainty about parameter estimates given v short follow up, and also to test the influence and effects of various parameters. Phase III trial – model used to draw cost-effectiveness conclusions and assess uncertainty around findings. 20 year time horizon. 81
 patient pathway & associated costs and outcomes beyond 28 days to identify parameters with the potential to be key drivers of cost-effectiveness. Use trial data, and other sources such as audit of AUGIB. The same model will be used to model costs/ effects beyond 28 days to identify key cost drivers as part of the feasibility trial, as will be used to extrapolate costs and effects beyond the end of the trial time horizon in the phase III trial. But, models being used in different ways. Feasibility – lots of sensitivity analysis as lot of uncertainty about parameter estimates given v short follow up, and also to test the influence and effects of various parameters. Phase III trial – model used to draw cost-effectiveness conclusions and assess uncertainty around findings. 20 year time horizon. 81.")
82
Summary This feasibility trial will allow us to
Determine workable data collection methods for resource use, unit costs and outcomes (quality of life) for the phase III trial Identify key parameters for focussed measurement in the phase III trial 82
 for the phase III trial. Identify key parameters for focussed measurement in the phase III trial. 82.")
83
Data Collection Forms in TRIGGER
All data collection is integrated into the CRFs Form 15 - length of stay around various departments and wards of hospital Follow up telephone contact at day 28 Form 12a – EQ-5D Forms 13a-f – resource use GP verification of patient reported events E.g. Form 5 captures medications, Form 8 endoscopies, Form 10b – # lab tests, # other blood products. Form 1a and 14 capture where patients were admitted from and discharged to. Forms particularly for health economics: Form 15 – length of stay around various depts and wards of hospital – clearly an important cost driver Telephone follow up 83

84
The Do’s and Dont’s of CRF Completion
Tania Reed Clinical Data Manager NHSBT 14:40-14:55

85
(Principle 11 - Records, WHO Good Clinical Research Practice)
All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation, and verification. (Principle Records, WHO Good Clinical Research Practice) Preparation and maintenance of clinical trial documents that leaves an audit trail throughout the conduct of the clinical trial.
 Preparation and maintenance of clinical trial documents that leaves an audit trail throughout the conduct of the clinical trial.")
86
The Importance of Data Quality and Integrity in Clinical Trials
Data quality refers to the extent to which data is accurate, legible, complete, original, and attributable to the person generating it Data integrity refers to the extent to which data is credible, consistent and verifiable It is important that we maximise data quality and integrity when recording information on CRFs and resolving data queries, in the TRIGGER trial Data quality and integrity both important when recording information on CRFs or resolving data queries.

87
Objectives of Clinical Data Management
To ensure the quality and integrity of clinical trial data To ensure that trial data is collected in accordance with the clinical trial protocol To provide a data set that is of sufficient quality for statistical analysis, interpretation and reporting To work in accordance with ICH-GCP principles International Conference on Harmonisation Good clinical practice principles

88
Things to remember when recording information on CRFs:
Write in black ink If you make an error put a make sure your original entry is visible, write your correction alongside, sign and date Do not scribble over your original entry Do not change numbers by writing over them Do not use correction fluid Do not destroy CRFs if you make an error(s)
")
89
Things to remember when recording information on CRFs:
If you make an error effecting the whole CRF page, put a line across the page, write ‘In error’, sign and date. Make sure the original entry is visible. Keep the copy in the patients file Write clearly, make sure it is legible Make sure that your writing is readable on fax or scanned copies

90
Things to remember when recording information on CRFs:
If data is unknown or not recorded write ‘UNK’ or ‘NR’ in boxes or write ‘UNKNOWN’ or ‘NOT RECORDED', alongside, sign and date When transcribing from source documents e.g. patient records, laboratory records, double check what you have written Include leading zeros e.g. write 8.23 as 08.23 Complete decimal places if boxes are provided for them, if no decimal place enter zero e.g. 4 should be 4.0

91
Things to remember when recording information on CRFs:
Use dd-mm-yyyy format for dates, if there are no boxes provided Use hh:mm format for times Double check your calculations before recording on CRF e.g. Blatchford score, Rockall score If you add any additional comments to CRFs remember to sign and date each one If sending any additional pages or documents with CRFs - include the patient’s trial no, date of birth and initials on each page

92
Sending CRFs to the CSU:
Check that all questions on the CRF have been completed Check that the patient’s trial no, initials and date of birth are correct and the same on all CRF pages Check that you have printed your name, signed and dated at the bottom of all CRF pages Do not write any patient identifiable information on any documents to be sent to the CSU

93
Sending CRFs to the CSU:
Always use a CRF transmittal form Send copies of the Screening Log every Monday Send CRFs in 3 batches for each patient (1) Trial entry (Forms 1a – 4) (2) Discharge or Day 28 (Forms 5a – 10b) (3) End of study/Follow up (Forms 11a – 14 & 18) Always complete a safety event transmittal form when sending Serious Adverse Events and Serious Transfusion Related Adverse Event CRFs
 Trial entry (Forms 1a – 4) (2) Discharge or Day 28 (Forms 5a – 10b) (3) End of study/Follow up (Forms 11a – 14 & 18) Always complete a safety event transmittal form when sending Serious Adverse Events and Serious Transfusion Related Adverse Event CRFs.")
94
Sending CRFs to the CSU:
The preferred method of sending the CRFs is by scanning and ing or by post or fax SAE forms At the CSU the Trial data manager will send an to confirm that the CRF pages or Screening log pages have been received

95
Resolving Data Queries:
Data Queries are usually raised due to:- Missing data Missing CRF pages Data anomalies The Trial Data Manager will send an to site staff, with details of the query

96
Resolving Data Queries:
Data clarification forms are generated by the database during data entry, and will be attached to the query as a pdf Data clarification form specifies the site, patient, form, question and the actual query text Site staff should clarify the discrepancy, provide missing data or missing CRF(s) Record the amended or missing data on the CRF Write the requested information in the ‘Answer’ section below the query text Show e.g. of data clarification form
 Record the amended or missing data on the CRF. Write the requested information in the ‘Answer’ section below the query text. Show e.g. of data clarification form.")
97
Resolving Data Queries:
If no data clarification form send an back responding to the query and attach or send a copy of the amended CRF Print your name, sign and date each data clarification form Send a copy of the amended CRF and/or missing CRF pages to CSU Keep the original amended CRF(s), and data clarification form in the site file Data query
, and data clarification form in the site file. Data query.")
98
Summary Discussed best practice when completing CRFs and resolving data queries The aim of following this best practice is to maximise data quality and integrity during the TRIGGER trial Reduce the number of data queries sent to you - thereby reducing your workload!

99
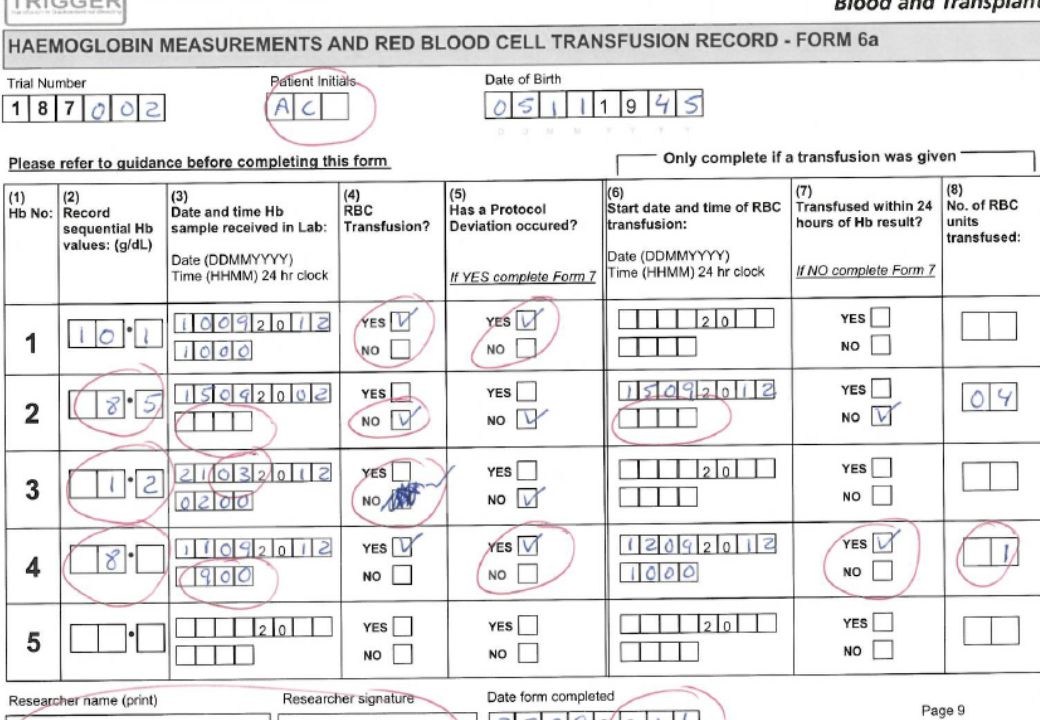
Exercise in pairs How many discrepancies can you find on this CRF - Haemoglobin Measurements and RBC Transfusion Record – Form 6a? We have completed Form 6a including quite a lot of errors ... We want you to try to find as many as you can...Work in pairs circle each error that you find... Once all finished run through the errors and give a sheet with how it should be completed...Give example of a data clarification form!!

106
Tea 14:55-15:10

107
Trial Site File Ana Mora 15:10-15:20

108
Investigator Site File
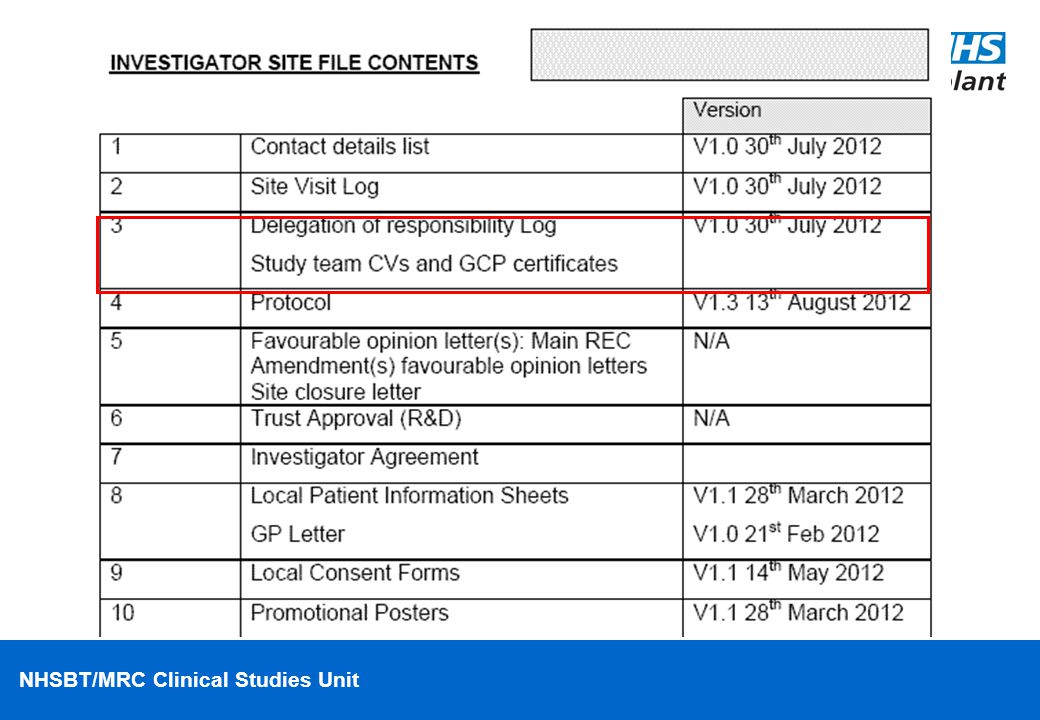
An Investigator Site File (ISF) contains essential documents on the trial and forms/documents used by the individual site • The responsibility to hold and maintain an up to date ISF, including all superseded documents, is with the Principal Investigator at each participating site When new or amended trial documents become available (e.g. protocols, PIS, reports, etc), it is the responsibility of the Trial coordinating team to provide copies to the Investigator Site together with relevant Ethics approvals
 contains essential documents on the trial and forms/documents used by the individual site. • The responsibility to hold and maintain an up to date ISF, including all superseded documents, is with the Principal Investigator at each participating site. When new or amended trial documents become available (e.g. protocols, PIS, reports, etc), it is the responsibility of the Trial coordinating team to provide copies to the Investigator Site together with relevant Ethics approvals.")
113
Site Initiation/Activation
Before enrolling patients each site must ensure they have: An Investigator Site file Written permission from the site R&D A signed study site agreement A signed delegation log.

114
Vipul Jairath, Ana Mora, Elizabeth Stokes, Tania Reed, Alison Deary
Open Forum for Q’s&A’s Vipul Jairath, Ana Mora, Elizabeth Stokes, Tania Reed, Alison Deary 15:20-16:20

115
Lower Gastrointestinal Bleeding
Pragmatic study – patient will have been included on the basis of a suspected UGIB which later turns out to be a LGIB. Stop completing any further CRFs and proceed to end of study form (Form 14) and indicate the patient did NOT complete the study as planned with the reason. Do not complete Day 28 follow up questionnaire Do return all completed CRFs and end of study form
 and indicate the patient did NOT complete the study as planned with the reason. Do not complete Day 28 follow up questionnaire. Do return all completed CRFs and end of study form.")
116
Co-Enrolment Guidelines
The only competing study we are currently aware of is the early TIPSS study in Edinburgh and Glasgow. Patients can be co-enrolled to this study. Any future competing studies will be considered on a case by case level after discussion with each trial steering committee.

117
Trial Website and Close
Vipul Jairath & Ana Mora 16:20-16:30

Similar presentations













 for Investigators and the Research Team.>")
 Changes Iowa Medicaid Enterprise October 14, 2008.>")








 WELCOME Support Workshop Thursday 7 th February 2013 1.>")

 Electronic Document Access (EDA)>")




