Download presentation
Presentation is loading. Please wait.

1
Molecular neurology Parkinson's disease
for image Matthias Georg Ziller R4 neurology January 30th 2008

2
Parkinson's disease Clinical picture first described in 1817
Motor features Resting tremor Bradykinesia Rigidity Postural instability Non motor features Autonomic Cognitive psychiatric

3
Burden of Parkinson's disease
~ patients in US 1 % of > 50 yo. 2nd most common MD Lifetime risk 2 % for men, 1.3 % for women Projected increase Patient numbers will double to 8-9 million worldwide in 2030 Dorsey et al., NEUROLOGY 2007;68: Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030

4
PD: A history of understanding
For a long time assumed to be a non hereditary disorder … but : 140 years ago – description of early familial cases 90 years ago – link between parkinsonism and cases of encephalitis and formation of inclusion bodies was exlored 45 years ago – demonstration of dopamine deficiency, discovery of nigro-striatal pathway and first symptomatic use of dopamine 25 years ago – observation of MPTP-induced toxicology 10 years ago – The molecular genetic revolution … monogenic forms … a "Nature versus nurture" debate

5
Pathophysiology of motor manifestations
D1 Result of the direct circuit is activation of cortex and facilitation of movement

6
Pathophysiology of motor manifestations
D2 Result of the indirect circuit is inhibition of thalamus, decreased activation of cortex and inhibition of movement – Action of SNpc is antagonistic to this

7
Pathophysiology of motor manifestations
Loss of dopaminergic stimulation of striatum results in greater inhibition of thalamus and decreased cortical activation bradykinesia

8
Neuropathology Loss of dopaminergic pigmented neurons in substantia nigra pars compacta

9
Neuropathology Cell loss in other pigmented nuclei
Ventral tegmental area Locus coeruleus Raphe nucleus Intraneuronal eosinophilic inclusion bodies (Lewy) Brainstem Diffuse distribution in cortex Staining + for alpha-synuclein Reactive gliosis
 Brainstem. Diffuse distribution in cortex. Staining + for alpha-synuclein. Reactive gliosis.")
10
Lewy body and Lewy neurites
-SYNUCLEINE +

11
Staging of PD pathology
2003 Braak et al.: Staging of PD pathology Allows to understand presymptomatic disease and non motor manifestations Inclusion bodies ascend in the brainstem Stage 2: coeruleus Sleep, mood At stage 3 reach SNpc Motor manifestations > stage 4 cortex involved Dementia Behavioural symptoms

12
Progression of a-synuclein immunopositive
labelling from stages 3 to 6, Braak 2006 Figure 3. a-d: Stages 3 to 6 of pathological changes associated with sporadic Parkinson's disease (PD) in 100 m sections immunostained for -synuclein (syn-1, Transduction Laboratories). The darkened tissue areas indicate the presence of PD-related lesions that are already recognizable with the naked eye from stage 3 onward in hemisphere sections. a: Hemisphere section of a case at stage 3. The arrow points to a single spot showing the central subnucleus of the amygdala. Allocortical, mesocortical, and neocortical areas are uninvolved at this stage. A close-up of the affected region appears in Figure 2e. b: Hemisphere section of a stage 4 case. More severe involvement of the amygdala (arrow) is accompanied by beginning affection of the anteromedial temporal mesocortex (arrowhead). Note that neocortical areas are not immunolabeled. c: Hemisphere section from a stage 5 case. A thick network of LNs emerges in the superficial layers of the anteromedial temporal mesocortex, and projection neurons located in the deep layers contain LBs (arrowhead). The disease process encroaches upon the related insular and cingulate mesocortex (asterisks). From the mesocortex, the pathology progresses into the high order association fields of the neocortex. The immunoreactivity gradually tapers off the closer it gets to the secondary and primary fields of the temporal neocortex (arrow). Note that the gyrus of Heschl along the superior temporal convolution is unaffected. d: Cortical involvement gains additional momentum in stage 6. Areas of the insular, cingulate (asterisks), and temporal mesocortex (arrowhead) continue to show strong immunolabeling. The cortical changes increase both in severity and extent. The disease process reaches even the secondary and, in advanced stage 6 cases, primary neocortical fields, as seen here from the mild affection of the primary auditory field in Heschl's gyrus (arrow).
 in 100 m sections immunostained for -synuclein (syn-1, Transduction Laboratories). The darkened tissue areas indicate the presence of PD-related lesions that are already recognizable with the naked eye from stage 3 onward in hemisphere sections. a: Hemisphere section of a case at stage 3. The arrow points to a single spot showing the central subnucleus of the amygdala. Allocortical, mesocortical, and neocortical areas are uninvolved at this stage. A close-up of the affected region appears in Figure 2e. b: Hemisphere section of a stage 4 case. More severe involvement of the amygdala (arrow) is accompanied by beginning affection of the anteromedial temporal mesocortex (arrowhead). Note that neocortical areas are not immunolabeled. c: Hemisphere section from a stage 5 case. A thick network of LNs emerges in the superficial layers of the anteromedial temporal mesocortex, and projection neurons located in the deep layers contain LBs (arrowhead). The disease process encroaches upon the related insular and cingulate mesocortex (asterisks). From the mesocortex, the pathology progresses into the high order association fields of the neocortex. The immunoreactivity gradually tapers off the closer it gets to the secondary and primary fields of the temporal neocortex (arrow). Note that the gyrus of Heschl along the superior temporal convolution is unaffected. d: Cortical involvement gains additional momentum in stage 6. Areas of the insular, cingulate (asterisks), and temporal mesocortex (arrowhead) continue to show strong immunolabeling. The cortical changes increase both in severity and extent. The disease process reaches even the secondary and, in advanced stage 6 cases, primary neocortical fields, as seen here from the mild affection of the primary auditory field in Heschl s gyrus (arrow).")
13
"Nurture": environmental contribution
Factors that increase the risk • Influenza epidemic - van Economo’s disease • Being raised on a farm with well water • Pesticide / herbicide / manganese exposure • MPTP toxin exposure Factors that modify (maybe decrease) the risk • Regular tobacco use and recent smoking • Caffeinated beverages (but not EtOH) • Physical exercise • Some NSAIDs (Ibuprofen, but not ASA) • Increased risk with high dairy intake in men • Higher levels of uric acid in peripheral blood
 the risk. • Regular tobacco use and recent smoking. • Caffeinated beverages (but not EtOH) • Physical exercise. • Some NSAIDs (Ibuprofen, but not ASA) • Increased risk with high dairy intake in men. • Higher levels of uric acid in peripheral blood.")
14
"Nature": Genetic forms Discovered during past 10 years
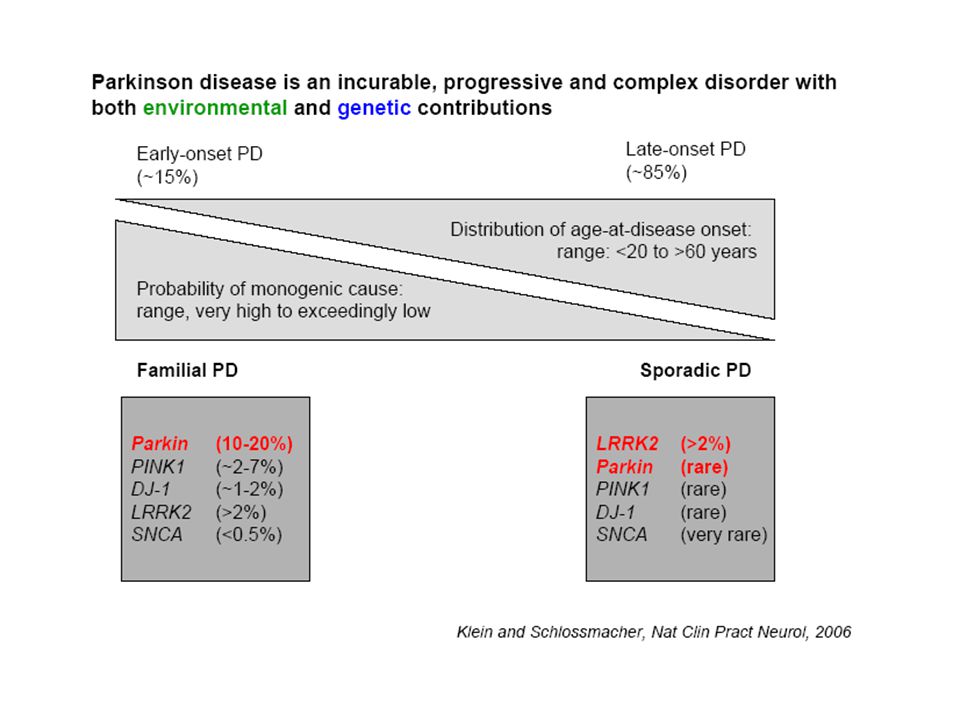
Monogenic forms may be clinically indistinguishable from "sporadic PD" 2-3 % of sporadic cases probably monogenic May lead to understanding of pathophysiology

15
Genetic forms

16
Familial forms - Summary
PARK 1-13 5 autosomal dominant: PARK 1 (=4), 4 is the triplication form of PARK1 3, 5, 8, 13 4 recessive: PARK 2,6,7,9 1 X-linked: PARK 12 2 with unknown mode of transmission: 10,11 additional mutations in small families without assigned PARK-numbers
, 4 is the triplication form of PARK1. 3, 5, 8, recessive: PARK 2,6,7,9. 1 X-linked: PARK with unknown mode of transmission: 10,11. additional mutations in small families without assigned PARK-numbers.")
17
Alpha-synuclein (PARK1+4)
First described + associated with familial parkinsonism 1997 SNCA gene, chrom 4q 3 known point mutations single-allele duplication or triplication in some families initially named PARK4 Penetrance low, 33 % Severity depends on gene dosage, patients with duplication resemble “idiopathic” PD more than triplication cases Mutations and multiplications are rare Increased expression of wild type SNCA copies may play a role in "idiopathic disease" First described + associated with familial parkinsonism 1997 SNCA gene, chrom 4q 3 known point mutations single-allele duplication or triplication in some families initially named PARK4 Penetrance low, 33 % Severity depends on gene dosage, patients with duplication resemble “idiopathic” PD more than triplication cases Broad spectrum of phenotypes Mutations and multiplications are rare Increased expression of wild type SNCA copies may play a role in "idiopathic disease"

18
What is SNCA ? 140-residue cytosolic and lipid-binding protein
Natively unfolded, presynaptic protein synaptic vesicle recycling, storage of transmitters negative coregulator of transmitter release Identical with Non-amyloid β component precursor Oligomer-forming SNCA in LBs truncated, oxidized and phosphorylated variants Mutants have greater nuclear targeting in cell culture What does it do ? has propensity to aggregate due to hydrophobic middle region, presence of fibrillar snca in LB, truncated forms mediate neurodegeneration in vivo in flies, mice aggregation is promoted by phosphorylation at Serin 129 and Serin-129-P is major component of LB role of G-protein coupled receptor kinase 5 is promoting aggregation Sept4 insuffiency (presynaptic scaffold protein) enhances Ser-129-P SNCA aggregation Unclear if protein accumulation is toxic or protective Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP SNCA may modulate oxidative damage (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity role for beta-SNC: blocks aggregation of aSNC and promotes cell survival
 enhances Ser-129-P SNCA aggregation. Unclear if protein accumulation is toxic or protective. Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity. Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking. Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP. SNCA may modulate oxidative damage. (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity. role for beta-SNC: blocks aggregation of aSNC and promotes cell survival.")
19
Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease
Figure. Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease. The binding of {alpha}-synuclein to lipid membranes protects it from misfolding and aggregation. Mutations in glucocerebrosidase (GBA) alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP SNCA may modulate oxidative damage (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity Feany M. N Engl J Med 2004;351:
 alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective. Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity. Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking. Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP. SNCA may modulate oxidative damage. (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity. Feany M. N Engl J Med 2004;351:")
20
LRRK-2 (PARK8) Leucin-rich repeat kinase 2 gene, identified by 2 groups in 2004 Mutations often associated with late-onset disease contrary to previous beliefs Large gene, 51 exons encoding AA- protein with several functional domains Certain genotypes may be susceptibility factors 16 sequence changes clearly pathogenic, in only 10 of 51 exons clustering in C-terminal region of protein pathology linked to kinase function but unclear* most frequent mutation is c.6055G A, 1.5 % of index cases with late-onset “idiopathic” PD same mutation found in 40% of Arab PD patients and 20 % of Ashkenazi Jewish PD likely founder effect homozygous forms of this mutation exist, but are not more severe than heterozygous Leucin-rich repeat kinase 2 gene, identified by 2 groups in 2004 Mutations often associated with late-onset disease contrary to previous beliefs Large gene, 51 exons encoding AA- protein with several functional domains, more than 50 known variants Certain genotypes may be susceptibility factors rather than pathogenic 16 sequence changes at least are clearly pathogenic, in only 10 of 51 exons clustering in C-terminal region of protein most frequent mutation is c.6055G A, 1.5 % of index cases with lat-onset “idiopathic” PD same mutation found in 40% of Arab PD patients and 20 % of Ashkenazi Jewish PD likely founder effect homozygous forms of this mutation exist, but are not more severe than heterozygous *Greggio et al., Neurobiol Dis 2006, Kinase activity is required for the toxic effects of mutant LRRK2/dardarin

21
Parkin (PARK2) Identified 1998, earlier age at onset, slower progression most common factor (10-20%) for early onset disease, all ethnic groups large number of known mutations 12 exons, chrom 6 Gene product Parkin: 465 aa protein, ubiquitin ligase, activity disrupted in mutants Mediates ubiquitilation of target proteins loss of ligase activity leads to pathology May lead to accumulation of substrates Parkin essential in formation of Lewy bodies ? Possible role in mitochondrial integrity Effector mechanisms Rescues mitochondrial dysfunction in flies after inactivation of PINK SNCA induced dysfunction is enhanced due to lack of Parkin activity Posttranslational modification of Parkin due to oxidative or nitrosative stress compromises ligase activity Parkin (PARK2) 1998, earlier age at onset, slower progression, better response to L-Dopa 2/6 autopsy brains had Lewy inclusions, not other 4 most common factor (10-20%) for early onset parkinsonism, all ethnic groups large number of known mutations, including small mutations and exon rearrangements 12 exons, chrom 6 gene product Parkin: 465 aa protein, ubiquitin ligase, activity disrupted in mutants mediates ubiquitilation of target proteins: targeting misfolded proteins to ubiquitin-proteasome pathway for degradation, loss of ligase activity leads to pathology ? Parkin essential in formation of Lewy bodies possible role in mitochondrial integrity Effector mechanisms Activation of IkappaB/NF-kB pathway by Parkin Rescues mitochondrial dysfunction in flies after inactivation of PINK SNCA induced dysfunction is enhanced due to lack of Parkin activity Posttranslational modification of Parkin due to oxidative or nitrosative stress compromises ligase activity The Parkin gene is expressed in presynaptic and postsynaptic processes and cell bodies of many neurons.19 The Parkin protein is presumed to function as an E3-type, E2-enzyme-dependent ubiquitin ligase that is involved in the proteasomal degradation of target proteins.20 The available E3 activity is disrupted by mutations associated with PD, thereby supporting the predominant loss-of-function theory.21 A growing number of putative ('to-be-ubiquitinated') Parkin substrates have been identified, and accumulation of these proteins is proposed to cause the selective death of neurons in the substantia nigra and locus coeruleus in humans.22 Although these putative substrates await convincing validation in animal models with disrupted parkin alleles, Drosophila melanogaster and mice that are deficient of wild-type parkin revealed unequivocal, systemic signs of increased oxidative stress and mitochondrial dysfunction.23, 24, 25 It is currently unclear if these biochemical changes can be attributed solely to the E3-ligase activity of the protein (or lack thereof) or to an—as yet unknown—function of the parkin homologs. An equally contentious issue is whether Parkin expression is essential for formation of neuronal inclusions in vivo, and if so, whether its ubiquitin ligase activity is responsible for the formation of Lewy bodies, as suggested (but not proven) by the abundance of ubiquitinated proteins in Lewy inclusions isolated from human brain.19 Disappointingly, the pathognomonic loss of human substantia nigra neurons has not been replicated in any of the published mouse models of three monogenic PD variants ( -synuclein [SNCA]-transgenic mice, parkin-null mice and dj1-null mice). By contrast, the fruit fly (D. melanogaster) appears more susceptible to PD-type pathology.26 Recently, two biochemical modifications of Parkin (S-nitrosylation and dopamine quinone-adduct formation) were identified in cellular studies and human brain specimens.27 These data indicated that reduced E3-ligase activity of the wild-type Parkin protein (rather than an autosomal recessive mutation in the two Parkin alleles) could also occur as a result of the principal pathogenetic process that is responsible for the development of sporadic PD.
 1998, earlier age at onset, slower progression, better response to L-Dopa. 2/6 autopsy brains had Lewy inclusions, not other 4. most common factor (10-20%) for early onset parkinsonism, all ethnic groups. large number of known mutations, including small mutations and exon rearrangements. 12 exons, chrom 6. gene product Parkin: 465 aa protein, ubiquitin ligase, activity disrupted in mutants. mediates ubiquitilation of target proteins: targeting misfolded proteins to ubiquitin-proteasome pathway for degradation, loss of ligase activity leads to pathology. Parkin essential in formation of Lewy bodies. possible role in mitochondrial integrity. Effector mechanisms. Activation of IkappaB/NF-kB pathway by Parkin. Rescues mitochondrial dysfunction in flies after inactivation of PINK. SNCA induced dysfunction is enhanced due to lack of Parkin activity. Posttranslational modification of Parkin due to oxidative or nitrosative stress compromises ligase activity. The Parkin gene is expressed in presynaptic and postsynaptic processes and cell bodies of many neurons.19 The Parkin protein is presumed to function as an E3-type, E2-enzyme-dependent ubiquitin ligase that is involved in the proteasomal degradation of target proteins.20 The available E3 activity is disrupted by mutations associated with PD, thereby supporting the predominant loss-of-function theory.21 A growing number of putative ( to-be-ubiquitinated ) Parkin substrates have been identified, and accumulation of these proteins is proposed to cause the selective death of neurons in the substantia nigra and locus coeruleus in humans.22 Although these putative substrates await convincing validation in animal models with disrupted parkin alleles, Drosophila melanogaster and mice that are deficient of wild-type parkin revealed unequivocal, systemic signs of increased oxidative stress and mitochondrial dysfunction.23, 24, 25 It is currently unclear if these biochemical changes can be attributed solely to the E3-ligase activity of the protein (or lack thereof) or to an—as yet unknown—function of the parkin homologs. An equally contentious issue is whether Parkin expression is essential for formation of neuronal inclusions in vivo, and if so, whether its ubiquitin ligase activity is responsible for the formation of Lewy bodies, as suggested (but not proven) by the abundance of ubiquitinated proteins in Lewy inclusions isolated from human brain.19 Disappointingly, the pathognomonic loss of human substantia nigra neurons has not been replicated in any of the published mouse models of three monogenic PD variants ( -synuclein [SNCA]-transgenic mice, parkin-null mice and dj1-null mice). By contrast, the fruit fly (D. melanogaster) appears more susceptible to PD-type pathology.26. Recently, two biochemical modifications of Parkin (S-nitrosylation and dopamine quinone-adduct formation) were identified in cellular studies and human brain specimens.27 These data indicated that reduced E3-ligase activity of the wild-type Parkin protein (rather than an autosomal recessive mutation in the two Parkin alleles) could also occur as a result of the principal pathogenetic process that is responsible for the development of sporadic PD.")
22
Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease
Figure. Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease. The binding of {alpha}-synuclein to lipid membranes protects it from misfolding and aggregation. Mutations in glucocerebrosidase (GBA) alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP SNCA may modulate oxidative damage (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity Feany M. N Engl J Med 2004;351:
 alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective. Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity. Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking. Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP. SNCA may modulate oxidative damage. (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity. Feany M. N Engl J Med 2004;351:")
23
Ubiquitin Proteasome System

24
Ubiquitine-Proteasome System in PD
Parkin, E3-UB ligase Loss of function in mutants Mediates ubiquitylation of synphilin, SNCA-interacting protein UCHL-1 (PARK5), deubiquitylating enzyme Impaired UB-hydrolysis Alpha-synuclein inhibits proteasome But no generalized dysfunction in brain regions with LBs Precise mechanism of UPS dysfunction in PD unclear Decreased function with toxin exposure ? MPTP inhibits proteasome Changes linked to mitochondrial function, aging ?
, deubiquitylating enzyme. Impaired UB-hydrolysis. Alpha-synuclein inhibits proteasome. But no generalized dysfunction in brain regions with LBs. Precise mechanism of UPS dysfunction in PD unclear. Decreased function with toxin exposure MPTP inhibits proteasome. Changes linked to mitochondrial function, aging")
25
PINK1(PARK6) =PTEN-induced kinase 1, mutations found in 2004
3 families with consanguinity and autosomal recessive parkinsonism 1-8 % of populations, considerable variation between ethnic groups Mutations near kinase domain loss of function in vivo 581 aa-protein, mitochondrial localization Highly conserved kinase domain, mitochondrial targeting motif no known substrate for kinase activity Mutations lead to different phosphorylation patterns Proteasomal stress enables altered cleavage of PINK and possible accumulation in LBs Interaction with DJ-1: recruited to pathway after oxidative damage due to PINK dysfunction

26
DJ-1 (PARK7) Described 2003, chrom 1, 1-2 % of early onset cases
Localization: ubiquitous expression cytosolic, may translocate to mitochondria with pH changes Function: chaperone-like activity, intracellular sensor of oxidative stress regulates D2 receptor signaling Direct action as antioxidant ? DJ-1 mutants show increased vulnerability to energy metabolism changes Overexpression of DJ-1 protects against mitochondrial complex 1 inhibitors effect is abrogated by mutants

27
Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease
Figure. Speculative Model of the Interactions among Proteins Implicated in Parkinson's Disease. The binding of {alpha}-synuclein to lipid membranes protects it from misfolding and aggregation. Mutations in glucocerebrosidase (GBA) alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP SNCA may modulate oxidative damage (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity Feany M. N Engl J Med 2004;351:
 alter lipid composition to favor the accumulation of cytosolic {alpha}-synuclein that can then misfold and aggregate into Lewy bodies. Mutations in DJ-1 may enhance the misfolding and aggregation of {alpha}-synuclein and other substrates. Mutations in parkin and UCH-L1 impair the ability of the cell to clear abnormal proteins. The misfolding and aggregation of {alpha}-synuclein and possibly other proteins trigger oxidative stress and energy depletion. PINK1 mutations compromise mitochondrial function, and the response to oxidative damage relies on normal DJ-1 function. A-synuclein: Unclear if protein accumulation is toxic or protective. Pharmacologic agents promoting inclusions formation seem to protect against SNCA toxicity. Early events after SNCA accumulation: blockade of ER to Golgi vesicular trafficking. Mice expressing human mutant SNCA develop mitochondrial pathology and SN neurons are vulnerable to MPTP. SNCA may modulate oxidative damage. (mice lacking SNCA are resistant to mitochondrial toxins) biochemical abnormalities in SNCA activate stress-signaling kinases, affect age-related decrease in neurogenesis, inhibit histone acetylation in nucleus to promote toxicity. Feany M. N Engl J Med 2004;351:")
28
Oxidative stress and mitochondrial dysfunction
Mitochondrial complex 1 inhibitors MPTP rotenone, paraquat Reproduce parkinsonism with dopaminergic neuron loss Absence of LB in these cases Chronic infusion reproduces all features, including LBs Supports theory of environmental toxin interaction by inhibition respiratory chain Inhibition of complex I: Depletion of ATP, impairment of dependent processes Generation of free radicals causing oxidative stress Post-mortem evidence of ox stress in PD brains Elevated lipid peroxidation and nitration levels in SN and LBs Reduced levels of glutathione and oxidized glutathione (antioxidants) Reduced complex 1 activity in muscle, brain, platelets of PD patients
 Reduced complex 1 activity in muscle, brain, platelets of PD patients.")
29
More questions: Role of susceptibility genes
Exact role is controversial: genes connected to monogenic forms may also act as susceptibility genes Single mutations in “recessive” genes are common (Parkin, DJ-1, PINK1) 2. Heterozygous mutations - may be associated with PET, MRI, ultrasound changes - Subtle motor signs in “asymptomatic” carriers - Unclear if these are developmental changes, early disease markers or adaptive 3. Are modifications of wild-type forms linked to parkinsonism ? ubiquitylation studies of wild-type Parkin linked to sporadic PD … 4. Genetic polymorphisms may be associated with disease - pG2385 increases risk in Chinese population
 2. Heterozygous mutations. - may be associated with PET, MRI, ultrasound changes. - Subtle motor signs in asymptomatic carriers. - Unclear if these are developmental changes, early disease. markers or adaptive. 3. Are modifications of wild-type forms linked to parkinsonism ubiquitylation studies of wild-type Parkin linked to sporadic PD … 4. Genetic polymorphisms may be associated with disease. - pG2385 increases risk in Chinese population.")
31
A model of known pathogenetic events in PD shows a principal imbalance between factors that promote PD (e.g. increased total metal content in the substantia nigra, altered steady-state levels of -synuclein proteins, including its phosphorylation, rise in dopamine-metabolism-related stress, and exposure to neurotoxins) and factors that prevent PD (e.g. cigarette smoking, caffeine consumption, expression of wild-type Parkin, DJ1, and PINK1, and normal levels of glutathione). LRRK2, leucine-rich repeat kinase 2; mt, mutant; phosphor., phosphorylation of -synuclein at residue Ser129; PINK1, phosphatase and tensin homolog (PTEN)-induced putative kinase 1; Ser18, serine at residue 18; Tyr, tyrosine at residue 18; UCHL1, ubiquitin carboxyl-terminal esterase L1; wt, wild-type.
 and factors that prevent PD (e.g. cigarette smoking, caffeine consumption, expression of wild-type Parkin, DJ1, and PINK1, and normal levels of glutathione). LRRK2, leucine-rich repeat kinase 2; mt, mutant; phosphor., phosphorylation of -synuclein at residue Ser129; PINK1, phosphatase and tensin homolog (PTEN)-induced putative kinase 1; Ser18, serine at residue 18; Tyr, tyrosine at residue 18; UCHL1, ubiquitin carboxyl-terminal esterase L1; wt, wild-type..")
32
References Continuum 2004, No.3 Movement Disorders, Suchowersky and Furtado Schlossmacher & Klein, Neurology, 2007, 69, 2093, PD – 10 years after its genetic revolution Lim et al, BMC Biochemistry 2007, 8, S1, Role of ubiquitin proteasome system in Parkinson's disease Thomas & Flint Beal, Parkinson's disease, Human Molecular Genetics 2007, Vol 16, Review Issue 2, Gandhi & Wood, Molecular pathogenesis of PD, Human Molecular Genetics 2005, 14, 18, Braak et al., Stanley Fahn Lecture: The staging procedure for Inclusion Body Pathology, Mov Disorders 21, no.12, 2006, Feany M. N Engl J Med 2004;351: 16th Annual National Residents’ Seminar on Movement Disorders Course Notes, Ottawa 2008

Similar presentations

 A 42 deposits subtle.>")




 Assis. Prof. Med. Sci. of Tehran Univ Dr. Pupak Derakhshandeh (PhD) Assis. Prof.>")



 -Pathology -Diagnosis - -synuclein.>")
 By Abdelkader Ashour, Ph.D. Phone: 4677212>")

 observations/experiences.>")




 -Pathology -Diagnosis - -synuclein.>")
