Download presentation
Presentation is loading. Please wait.

1
G53BIO – Bioinformatics http://www.cs.nott.ac.uk/~jqb/G53BIO
Protein Structure Prediction Dr. Jaume Bacardit – Some material taken from “Arthur Lesk Introduction to Bioinformatics 2nd edition Oxford University Press 2005” and “Introduction to Bioinformatics by Anna Tramontano”

2
Outline Introduction and motivation
Basic concepts of protein structure PSP: A family of problems Prediction of structural aspects of protein residues Prediction of the 3D structure of proteins Assessment of PSP quality: CASP Summary

3
Protein Structure: Introduction
Proteins are molecules of primary importance for the functioning of life Structural Proteins (collagen nails hair etc.) Enzymes Transmembrane proteins Proteins are polypeptide chains constructed by joining a certain kind of peptides amino acids in a linear way The chain of amino acids however folds to create very complex 3D structures There is a general consensus that the end state of the folding process depends on the amino acid composition of the chain
 Enzymes. Transmembrane proteins. Proteins are polypeptide chains constructed by joining a certain kind of peptides amino acids in a linear way. The chain of amino acids however folds to create very complex 3D structures. There is a general consensus that the end state of the folding process depends on the amino acid composition of the chain.")
4
Motivation for PSP The function of a protein depends greatly on its structure The structure that a protein adopts is vital to it’s chemistry Its structure determines which of its amino acids are exposed to carry out the protein’s function Its structure also determines what substrates it can react with However the structure of a protein is very difficult to determine experimentally and in some cases almost impossible

5
Protein Structure Prediction
That is why we have to predict it PSP aims to predict the 3D structure of a protein based on its primary sequence

6
Impact of PSP PSP is an open problem. The 3D structure depends on many variables It has been one of the main holy grails of computational biology for many decades Impact of having better protein structure models are countless Genetic therapy Synthesis of drugs for incurable diseases Improved crops Environmental remediation

7
Protein Structure

8
Backbone and side chain
All amino acids have a common part: the backbone Each amino acid type has a different side chain The Cα atom connects the backbone and the side chain The first carbon atom in the side chain is called Cβ (except for Gly)
")
9
Amino Acids

10
Protein Structure: Introduction
Different amino acids have different properties These properties will affect the protein structure and function Hydrophobicity, for instance, is the main driving force (but not the only one) of the folding process
 of the folding process.")
11
Protein Structure: Hierarchical nature of protein structure
Primary Structure = Sequence of amino acids MKYNNHDKIRDFIIIEAYMFRFKKKVKPEVDMTIKEFILLTYLFHQQENTLPFKKIVSDLCYKQSDLVQHIKVLVKHSYISKVRSKIDERNTYISISEEQREKIAERVTLFDQIIKQFNLADQSESQMIPKDSKEFLNLMMYTMYFKNIIKKHLTLSFVEFTILAIITSQNKNIVLLKDLIETIHHKYPQTVRALNNLKKQGYLIKERSTEDERKILIHMDDAQQDHAEQLLAQVNQLLADKDHLHLVFE Secondary Structure Tertiary Local Interactions Global Interactions

12
Protein Structure: Hierarchical nature of protein structure
The amino acid composition of a protein is called primary structure or primary sequence The folding process of a protein involves several steps The protein creates some patterns due to local interactions with the closest residues in the chain. These patters are called the protein secondary structure Afterwards, the secondary structure motifs organise into stable patters, called tertirary structure Finally, proteins can be composed of several subunits or monomers, forming the quaternary structure Other, less used, levels of this hierarchy are Supersecondary structure (recurrent patters of interaction between secondary structure elements close in sequence ) Domains (subunits within a protein with quasi-independent folding stability)
 Domains (subunits within a protein with quasi-independent folding stability)")
13
Backbone The polypeptide chain of proteins in joined together in a very specific way Two dihedral angles (phi and psi) define the torsion of each amino acid in the chain Phi is the angle of the Cα –N bond and psi is the angle of the Cα-C bond.
 define the torsion of each amino acid in the chain. Phi is the angle of the Cα –N bond and psi is the angle of the Cα-C bond.")
14
Protein Structure: Hierarchical nature of protein structure
Residues form a loop of 3.6 residues/turn and 5.4Å wide There are two main kinds of secondary structure motifs: α helices β sheets Residues that do not fail in these two categories are said to be in coil state Residues lay flat in parallell strands. Called parallell sheets if all strands have the same N-to-C orientation, and antiparallell if adjacent strands have opposed orientations

15
Protein Structure: Hierarchical nature of protein structure
Supersecondary structure elements β hairpin β-α-β unit

16
Protein Data Bank Proteins for which scientists have been able to resolve the structure (using x-ray crystallography, NMR, etc.) are stored in the Protein Data Bank (PDB) Each protein has a four letter ID code (PDB id) A fifth letter (A, B, C, etc.) is used to identify the chain within the protein Proteins are stored in a format called also PDB format File for the 1A68 protein
 are stored in the Protein Data Bank (PDB) Each protein has a four letter ID code (PDB id) A fifth letter (A, B, C, etc.) is used to identify the chain within the protein. Proteins are stored in a format called also PDB format. File for the 1A68 protein.")
17
Protein Structure: Ramachandran plots
We saw that the backbone of a residue is characterised by two angles: psi and phi. Can they take any value? Fortunately not This effect was studied long ago by GN Ramachandran He proposed a diagram to visualize these angles (phi in the X axis, psi in the Y axi) of amino acid residues Different types of secondary structure are clustered in different regions of the diagram
 of amino acid residues. Different types of secondary structure are clustered in different regions of the diagram.")
18
Protein Structure: Ramachandran plots
In real proteins, these plots are not so clear You can create the Ramachandran plot for any protein in PDB at At the right there is the plot for a set of 80 proteins

19
Protein Structure: Classifications of protein structure
Several tertiary structure classification method exists, for instance, SCOP, CATH, and FSSP/DDD. No method is perfect, hence was proposed. SCOP is the most widespread of them SCOP = Structural Classification Of Proteins In its 1.73 release (November 2007) it catalogs proteins with known structure (that is, entries in the PDB archive) It uses a hierarchical system to catalog the proteins, according to evolutionary origin and structural similarity The levels of the hierarchy are: class, fold, superfamily, family, protein and species
 it catalogs proteins with known structure (that is, entries in the PDB archive) It uses a hierarchical system to catalog the proteins, according to evolutionary origin and structural similarity. The levels of the hierarchy are: class, fold, superfamily, family, protein and species.")
20
Protein Structure: Classifications of protein structure
Main classes of SCOP (first level of hierarchy) All α proteins – proteins that have (almost) only α helices All β proteins – proteins that have (almost) only β sheets α+β proteins – proteins that have both α helices and (mostly) antiparallell strands, but segregated in different parts of the protein α/β proteins – proteins that have both α helices and (mostly) parallell strands, typically forming β+α+β units Multidomains proteins – proteins having two or more domains belonging to different classes Membrane and cell surface proteins Small proteins (metal ligans, heme and proteins with disulfide bridges Coiled coils proteins Low resolution protein structure Peptides Designed proteins
 All α proteins – proteins that have (almost) only α helices. All β proteins – proteins that have (almost) only β sheets. α+β proteins – proteins that have both α helices and (mostly) antiparallell strands, but segregated in different parts of the protein. α/β proteins – proteins that have both α helices and (mostly) parallell strands, typically forming β+α+β units. Multidomains proteins – proteins having two or more domains belonging to different classes. Membrane and cell surface proteins. Small proteins (metal ligans, heme and proteins with disulfide bridges. Coiled coils proteins. Low resolution protein structure. Peptides. Designed proteins.")
21
Protein Structure: Classifications of protein structure
SCOP classification of Flavodoxin from Clostridium beijerinckii Class: α/β Fold: Flavodoxin-like: 3 layers, α/β/α; parallel β-sheet of 5 strands Superfamily: Flavoproteins Family: Flavodoxin-related binds FMN Protein: Flavodoxin Species: Clostridium beijerinckii PDB ID: 5ULL

22
Prediction types of PSP
There are several kinds of prediction problems within the scope of PSP The main one of course is to predict the 3D coordinates of all atoms of a protein (or at least the backbone) based on its primary sequence There are many structural properties of individual residues within a protein that can be predicted for instance: The secondary structure state of the residue If a residue is buried in the core of the protein or exposed in the surface Accurate predictions of these sub-problems can simplify the general 3D PSP problem
 based on its primary sequence. There are many structural properties of individual residues within a protein that can be predicted for instance: The secondary structure state of the residue. If a residue is buried in the core of the protein or exposed in the surface. Accurate predictions of these sub-problems can simplify the general 3D PSP problem.")
23
Prediction types of PSP
There is an important distinction between the two classes of prediction The 3D PSP is generally treated as an optimisation problem The prediction of structural aspects of protein residues are generally treated as machine learning problems

24
Optimisation Given a problem for which you have a way of assessing how good is each possible solution An evaluation function Optimisation is the process of finding the best possible solution Dynamic programming (as seen for sequence alignment) is an optimisation method Genetic Algorithms are another examples of optimisation The key differences between them is how they explore the space of candidate solutions
 is an optimisation method. Genetic Algorithms are another examples of optimisation. The key differences between them is how they explore the space of candidate solutions.")
25
Machine Learning Machine learning: How to construct programs that automatically learn from experience [Mitchell 1997] ML is a Computer Science discipline part of the Artificial Intelligence field Its goal is to construct automatically a description of some phenomenon given a set of data extracted from previous observations of the phenomenon because it would be beneficial to predict it in the future.
![Machine Learning Machine learning: How to construct programs that automatically learn from experience [Mitchell 1997]](http://slideplayer.com/slide/2400751/8/images/25/Machine+Learning+Machine+learning%3A+How+to+construct+programs+that+automatically+learn+from+experience+%5BMitchell+1997%5D.jpg "ML is a Computer Science discipline part of the Artificial Intelligence field. Its goal is to construct automatically a description of some phenomenon given a set of data extracted from previous observations of the phenomenon because it would be beneficial to predict it in the future.")
26
Flow of data in machine learning
Specifically we are concerned with supervised learning. That is when we know the solution for the training data Training Set Learning Method Theory Unknown instance Class

27
Types of machine learning
Rule learning 1 If (X<0.25 and Y>0.75) or (X>0.75 and Y<0.25) then If (X>0.75 and Y>0.75) then If (X<0.25 and Y<0.25) then Y Everything else 1 X
 or. (X>0.75 and Y<0.25) then If (X>0.75 and Y>0.75) then If (X<0.25 and Y<0.25) then Y. Everything else 1. X.")
28
Other machine learning techniques
Other methods that have also been used in PSP are Artificial Neural Networks Support Vector Machines Hidden Markov Models If you are interested in the technology side of PSP a good book is “Bioinformatics: The Machine Learning Approach” by Baldi and Brunak

29
Prediction of structural aspects of protein residues
Many of these features are due to local interactions of an amino acid and its immediate neighbours Can it be predicted using information from the closest neighbours in the chain? In this simplified example to predict the SS state of residue i we would use information from residues i-1 i and i+1. That is a window of ±1 residues around the target Ri-5 SSi-5 Ri-4 SSi-4 Ri-3 SSi-3 Ri-2 SSi-2 Ri-1 SSi-1 Ri SSi Ri+1 SSi+1 Ri+2 SSi+2 Ri+3 SSi+3 Ri+4 SSi+4 Ri+5 SSi+5 Ri-1 Ri Ri+1 SSi Ri Ri+1 Ri+2 SSi+1 Ri+1 Ri+2 Ri+3 SSi+2

30
What information do we include for each residue?
Early prediction methods used just the primary sequence the AA types of the residues in the window However the primary sequence has limited amount of information It does not contain any evolutionary information it does not say which residues are conserved and which are not Where can we obtain this information? Position-Specific Scoring Matrices which is a product of a Multiple Sequence Alignment

31
Position-Specific Scoring Matrices (PSSM)
For each residue in the query sequence compute the distribution of amino acids of the corresponding residues in all aligned sequences (discarding those too similar to the query) This distributions will tell us which mutations are likely and which mutations are less likely for each residue in the query sequence In essence it’s similar to a substitution matrix but tailored for the sequence that we are aligning A PSSM profile will also tell us which residues are more conserved and which residues are more subject to insertions or deletions
 This distributions will tell us which mutations are likely and which mutations are less likely for each residue in the query sequence. In essence it’s similar to a substitution matrix but tailored for the sequence that we are aligning. A PSSM profile will also tell us which residues are more conserved and which residues are more subject to insertions or deletions.")
32
PSSM for the 10 first residues of 1n7lA
A R N D C Q E G H I L K M F P S T W Y V A: M: E: K: V: Q: Y: L: T: R:

33
Secondary Structure Prediction
The most usual way is to predict whether a residue belongs to an α helix a β sheet or is in coil state Several programs can determine the actual SS state of a protein from a PDB file. The most common of them is DSSP Typically, a window of ±7 amino acids (15 in total) is used
 is used.")
34
Secondary Structure Prediction
MSA R1 R2 R3 Rn-1 Rn PSSM1 PSSM2 PSSM3 PSSMn-1 PSSMn Primary sequence PSSM profile of sequence Prediction method Windows generation PSSMi-1 PSSMi PSSMi+1 SSi? Window of PSSM profiles Prediction The most popular public SS predictor is PSIPRED

35
Coordination Number Prediction
Two residues of a chain are said to be in contact if their distance is less than a certain threshold (e.g. 8Å) CN of a residue : count of contacts that a certain residue has CN gives us a simplified profile of the density of packing of the protein Native State Primary Sequence Contact
 CN of a residue : count of contacts that a certain residue has. CN gives us a simplified profile of the density of packing of the protein. Native State. Primary. Sequence. Contact.")
36
Other predictions Other kinds of residue structural aspects that can be predicted Solvent accessibility: Amount of surface of each residue that is exposed to solvent Recursive Convex Hull: A metric that models a protein as an onion and assigns each residue to a layer. Formally each layer is a convex hull of points These features (and others) are predicted in a similar was as done for SS or CN
 are predicted in a similar was as done for SS or CN.")
37
Contact Map prediction
Prediction given two residues from a chain whether these two residues are in contact or not This problem can be represented by a binary matrix. 1= contact 0 = non contact Plotting this matrix reveals many characteristics from the protein structure helices sheets

38
Contact Map Prediction
Instead of a single window around the target now there are two windows around the pair of residues to be predicted to be in contact or not Many methods also use a third window, placed in the middle point in the chain between the two target residues

39
Contact Map prediction at Nottingham
For each position in these 3 windows we include: PSSM profile Predicted SS, SA, RCH and CN The whole connecting segment between the two targets is represented as Distribution of AA and predicted SS, SA, RCH and CN

40
Contact Map prediction at Nottingham
Moreover, global protein information is also included Sequence length Separation between target residues Contact propensity of target residues Distribution of AA and predicted SS, SA, RCH and CN of the whole chain Each instance is represented by 631 variables

41
Contact Map prediction at Nottingham
Training set x50 x25 Consensus Predictions Samples Rule sets Training set of 2413 proteins selected to represent a broad set of sequences 32 million pairs of amino-acids (instances in the training set) with less than 2% of real contacts Each instance is characterized by up to 631 attributes 50 samples of ~ examples are generated from the training set. Each sample contains two no-contact instances for each contact instance The BioHEL GBML method (Bacardit et al., 2009) was run 25 times on each sample An ensemble of 1250 rule sets (50 samples x 25 seeds) performs the contact maps predictions using simple consensus voting Confidence is computed based on the votes distribution in the ensemble (Bacardit et al., Bioinformatics (2012) 28 (19): )
 with less than 2% of real contacts. Each instance is characterized by up to 631 attributes. 50 samples of ~ examples are generated from the training set. Each sample contains two no-contact instances for each contact instance. The BioHEL GBML method (Bacardit et al., 2009) was run 25 times on each sample. An ensemble of 1250 rule sets (50 samples x 25 seeds) performs the contact maps predictions using simple consensus voting. Confidence is computed based on the votes distribution in the ensemble. (Bacardit et al., Bioinformatics (2012) 28 (19): )")
42
3D Protein Structure Prediction
Approaches for 3D PSP Template-Based Modelling Ab-Initio methods State-of-the-Art methods I-Tasser Rosetta

43
Approaches for 3D PSP Some PSP methods try to identify a template protein and then adapt the structure of the template to the target protein Template-based Modelling Other methods try to generate the structure of the protein from scratch (Ab Initio Modelling) optimizing some energy function that models the stability of the protein, in case that no template can be identified
 optimizing some energy function that models the stability of the protein, in case that no template can be identified.")
44
Pipeline for Template-based Modelling
Typical steps Identify the template (next slide) Produce the final alignment between the residues of target and template Determine main chain segments to represent the regions containing insertions and deletions (gaps in the alignment) and stitch them into the main chain of the template to create an initial model for the target Replace the side chains of residues that have been mutated (mismatches in the alignment) although it is possible that the conformation in the template is still conserved Examine the model to detect any serious atom collision and relieve them Refine the model by energy minimization. This stage is meant to adapt the stitched segments to the conserved structure and to adjust the side chains so find the most stable conformation
 Produce the final alignment between the residues of target and template. Determine main chain segments to represent the regions containing insertions and deletions (gaps in the alignment) and stitch them into the main chain of the template to create an initial model for the target. Replace the side chains of residues that have been mutated (mismatches in the alignment) although it is possible that the conformation in the template is still conserved. Examine the model to detect any serious atom collision and relieve them. Refine the model by energy minimization. This stage is meant to adapt the stitched segments to the conserved structure and to adjust the side chains so find the most stable conformation.")
45
Loop remodelling

46
Template identification
Can we find a sequence with known structure and high sequence identify with the target? Homology Modelling Still, there is a template (structure similar to that of the target) but it has poor sequence identity. We need to identify it by other means Fold recognition Profile-based methods Threading methods
 but it has poor sequence identity. We need to identify it by other means. Fold recognition. Profile-based methods. Threading methods.")
47
Profile-based Methods
Aim is to construct 1D representations (profiles) of the structures in our fold database Afterwards, when a target sequence comes, we construct its profile and check our database for the most similar profile That is, instead of aligning amino acid sequences, we align structural 1D profiles
 of the structures in our fold database. Afterwards, when a target sequence comes, we construct its profile and check our database for the most similar profile. That is, instead of aligning amino acid sequences, we align structural 1D profiles.")
48
How to construct the profile?
We choose a series of structural properties of residues Most frequent secondary structure state Alpha helix, Beta sheet, other Solvent Accessibility < 40Å2, >100Å2, intermediate Hydrophobic/polar For each amino acid, we decide to which category it belongs based on statistics computed on a large database of structures

49
How to construct the profile?
Alpha helix Beta sheet Other <40Å2 Hydrophobic: a Polar: d Hydrophobic: b Polar: e Hydrophobic: c Polar: f >100Å2 Hydrophobic: g Polar: j Hydrophobic: h Polar: k Hydrophobic: i Polar: l intermediate Hydrophobic: m Polar: p Hydrophobic: n Polar: q Hydrophobic: o Polar: r Now the sequence for each protein in our database will have a new structural representation We need to predict SS and Acc for the template

50
Threading methods We start with compiling a catalogue of unique folds (filtering out repeats) Afterwards, we evaluate how likely it is that the target sequence adopts each of the folds, and how (alignment) Name is a metaphor taken from tailoring, as we are are trying to fit the sequence (a thread) through a known structure We will choose the template (and alignment) that has the lowest (estimated) energy
 Name is a metaphor taken from tailoring, as we are are trying to fit the sequence (a thread) through a known structure. We will choose the template (and alignment) that has the lowest (estimated) energy.")
51
Threading methods Energy estimation needs to be simple and fast
As we need to evaluate all possible folds and alignments Energy is the product of all the pair-wise interactions ocurring in a protein Thus, the energy estimation will be computed as the sum of the energy terms for every pair of residues in the protein How to compute the energy interaction for a given pair of amino acids?

52
Pair-wise Energy estimation
Boltzmann’s equation states that the probablity of observing a given event depends on its energy P(x) = e(E(x)/KT) If we reverse this equation we get: E(x) = -KT ln[ P(x) ] We can compute P(x), for each pair of amino acids from a database of known structures as the frequency in which these amino acids are observed to be in contact
 = e(E(x)/KT) If we reverse this equation we get: E(x) = -KT ln[ P(x) ] We can compute P(x), for each pair of amino acids from a database of known structures as the frequency in which these amino acids are observed to be in contact.")
53
Alignment within threading
We still need to solve the problem of the correspondence of the residues in our template with those of the target This is a very difficult problem, as a change in an alignment can have impact in the interaction with many residues There is an exact (but costly) solution Instead, most methods adopt an approximate method called frozen approximation When evaluating the possibility of assigning one of the amino acids of the target to a certain position in the template, instead of computing the interactions with the rest of the target residues, we will use those of the template
 solution. Instead, most methods adopt an approximate method called frozen approximation. When evaluating the possibility of assigning one of the amino acids of the target to a certain position in the template, instead of computing the interactions with the rest of the target residues, we will use those of the template.")
54
Frozen Apporximation

55
Aligning target and template
Crucial step before generating the initial model It is possible, specially for homology modelling, that the best sequence alignment does not correspond to the best structural alignment That is, finding the best correspondence between the coordinates of each amino acid of target and template In this case, a better alignment process needs to be performed, to do se, we can use Information derived from the template’s structure Predicted for the target

56
Aligning Target and Template
Wrong alignment. Some atoms are too close (big circle). Some atoms are too far (small circle) Correct alignment after shifting
. Some atoms. are too far (small circle) Correct alignment after shifting.")
57
The poor man approach to homology modelling
To find templates PSI-BLAST 3D Jury. This program is a meta-server. That is it asks many other servers what templates would they choose and then produces a consensus decision based on the answers of the servers To produce a model of a protein given a template MODELLER. Very popular homology modelling package. Free for academic use To refine the side-chain conformations SCWRL

58
Ab-Initio modelling In general this kind of modelling is still quite primitive when compared to homology modelling However without a target it is the only choice Pure ab-initio modelling is still very costly and ineffective but hybrid homology/ab-initio methods such as fragment assembly have better performance

59
Ab-Initio modelling The most advanced ab-initio method is fragment assembly Consists by breaking up the sequence in small subsegments of 3 to 9 residues and generating structure for these segments based on a large library of known fragments Decoys are generated from all possible combinations of fragments An energy minimization process is applied to all decoys. Decoys are clustered and the final models are selected from the center of the largest clusters

60
Energy minimisation Energy minimization is not easy. We may need to go uphill before we can reach the lowest energy conformation

61
Energy functions for ab-initio methods
Energy function needs to take into account the interactions of all atoms of all amino acids Many different types of energy sources Covalent bonds Angles and torsions of bonds between atoms Van der Waals interactions (repulsion/attraction) Energy of charged atoms Interactions with solvent Hydrogen bonds Exact formulas are very costly, so generally PSP methods use knowledge-based potentials, computed from a large database of structures
 Energy of charged atoms. Interactions with solvent. Hydrogen bonds. Exact formulas are very costly, so generally PSP methods use knowledge-based potentials, computed from a large database of structures.")
62
I-Tasser Prediction method from Zhang’s group
Fully automated server, without any human intervention Steps Template identification Structure assembly Atomic model construction Model selection

63
I-Tasser: Template Identification
MUSTER fold recognition method, used both for whole proteins (TBM) or for fragments (Ab Inition) Profile-based fold recognition Secondary structure Structural frament profile Solvent accessibility Backbone torsion angle Hydrophobicity For the most difficult targets, a meta-server that combines the outputs of various methods is used
 or for fragments (Ab Inition) Profile-based fold recognition. Secondary structure. Structural frament profile. Solvent accessibility. Backbone torsion angle. Hydrophobicity. For the most difficult targets, a meta-server that combines the outputs of various methods is used.")
64
I-Tasser: Structure assembly
Generation of a preliminary model with only coordinates for Cα and sidechain positions Using the template as starting point where possible and ab-initio methods for amino acids without alignment Two iterations of refinement 1st based on templates 2nd based on clustering the models of the previous iteration and using the centroids of each cluster as starting points

65
I-Tasser energy function
Knowledge-based statistics of Cα – sidechain correlation H-bonds Hydrophobicity Spatial restraints of templates Contact Map prediction from SVMSEQ 9 predictions included, combinations of Contacts between Cα, Cβ or side chain centers Contact cut-offs of 6, 7 or 8 Å

66
I-Tasser atomic model construction
Full-atom models are constructed from the approximate models produced by the cluster centroids 1st the backbone is matched with a large library of template fragments with high resolution structure Then full-atom optimization occurs focusing on H-bonds, removing clashes and using the Charmm22 molecular dynamics force field

67
I-Tasser model selection
Several full-atom models are generated from each cluster centroid Models need to be ranked to select the best one I-Tasser uses a weighted sum of Number of H-Bonds / target length TM-score (metric to compare structures) between the full-atom model and the centroid cluster
 between the full-atom model and the centroid cluster.")
68
Rosetta Predictor from David Baker’s group
It uses a massive distributed computing infrastructure For CASP7 in 2006 it claimed to dedicate up to 104 cpu years/target Template identification used a variety of methods depending on sequence identity between target and template Different protocols for Template-Based Modelling and Free Modelling (fragment assembly) 3 variants of TBM depending on degree of homology between target and template
 3 variants of TBM depending on degree of homology between target and template.")
69
Rosetta Full-atom refinement protocol Energy function based on
Short-range interations: Van der Waals energe, H-bonds and solvent accessibility Long range interactions (dampening of electrostatic interactions) Minimization through Monte Carlo with the following steps: Perturbation of a randomly selected angle from the backbone Optimisation of side-chain rotamer conformations Optimisation of both backbone and sidechain torsion angles
 Minimization through Monte Carlo with the following steps: Perturbation of a randomly selected angle from the backbone. Optimisation of side-chain rotamer conformations. Optimisation of both backbone and sidechain torsion angles.")
70
PSP and CASP PSP has improved through the years. This improvement has been assessed mainly in CASP CASP = Critical Assessment of Techniques for Protein Structure Prediction It is a biannual community exercise to evaluate the state-of-the-art in PSP Every day for about three months the organizers release some protein sequences for which nobody knows the structure (128 sequences were released in CASP8 in 2008) Each prediction group is given three weeks to return their predictions. 24 hours are give to automated servers Then at the end of the year experts meet in a place close to the sea to discuss the results of the experiment
 Each prediction group is given three weeks to return their predictions. 24 hours are give to automated servers. Then at the end of the year experts meet in a place close to the sea to discuss the results of the experiment ")
71
CASP categories Several categories of experiments are assessed in CASP
Template-Based Modeling (Homology and fold recognition) Free Modeling (no template i.e. ab initio) Contact Map prediction Functional sites prediction Domain prediction Disordered regions Quality assessment Categories have changed through time SS prediction is not assessed anymore after CASP4 Homology modeling and fold recognition merged into TBM
 Free Modeling (no template i.e. ab initio) Contact Map prediction. Functional sites prediction. Domain prediction. Disordered regions. Quality assessment. Categories have changed through time. SS prediction is not assessed anymore after CASP4. Homology modeling and fold recognition merged into TBM.")
72
Progress through CASP 1. Computers help structure prediction:
(From Nick Grishin’s Humans vs Servers presentation in CASP8) 1. Computers help structure prediction: no more paper models 2. Knowledge-based potentials work better. 3. Local “threading” and fragment assembly (Baker) 4. Averaging and consensus methods work: meta-servers (Ginalski-Rychlewski) 5. Sequence profile methods are as (or more powerful) than threading: (Sốding) 6. Jamming poorly similar templates together helps: (Skolnick-Zhang)
 1. Computers help structure prediction: no more paper models. 2. Knowledge-based potentials work better. 3. Local threading and fragment assembly. (Baker) 4. Averaging and consensus methods work: meta-servers (Ginalski-Rychlewski) 5. Sequence profile methods are as. (or more powerful) than threading: (Sốding) 6. Jamming poorly similar templates together. helps: (Skolnick-Zhang)")
73
Assessment of 3D PSP How can we quantify how good is a model?
That is, how similar is a model structure to the actual (native) one? We will see this in depth when we cover the protein structure comparison topic, later in the module Now we are just going to describe the most popular metric, GDT-TS
 one We will see this in depth when we cover the protein structure comparison topic, later in the module. Now we are just going to describe the most popular metric, GDT-TS.")
74
GDT-TS Global Distance Test – Total Score
This measure tries to produce a balance between good local and global similarity of structures (unlike RMSD) If a measure only takes a global point of view, good models that only fail badly in a few amino acids could be discarded
 If a measure only takes a global point of view, good models that only fail badly in a few amino acids could be discarded.")
75
GDT-TS steps All segments of 3, 5 and 7 consecutive amino acids from the model are superimposed to the actual structure. Each of them will be iteratively extended while they are good enough Good enough = Distance between all residue pairs (represented by their Cα atoms) is less than a certain threshold A final superposition includes the set of segments covering as many residues as possible Segments do not need to be continuous
 is less than a certain threshold. A final superposition includes the set of segments covering as many residues as possible. Segments do not need to be continuous.")
76
GDT-TS metric The process of superposition is performed four times, using thresholds of 1, 2, 4 and 8 Å The reason for including 4 different thresholds is to have a metric which is good both for high accuracy models and for approximate models

77
GDT-HA HA = High Accuracy
Set of thresholds in GDT-TS changed to 0.5, 1, 2 and 4 For high accuracy GDT just provide a crude approximation (backbone). So other measures are taken into account H-bonds Position and rotation of sidechains Clashes of atoms
. So other measures are taken into account. H-bonds. Position and rotation of sidechains. Clashes of atoms.")
78
Contact Map prediction in CASP
Contact Map is assessed using the targets in the Free Modelling category Also, only long-range contacts (with a minimum chain separation of 24 residues) are evaluated Predictor groups are asked to submit a list of predicted contacts and a confidence level for each prediction The assessors then rank the predictions for each protein and take a look at the top L/x ones, where L is the length of the protein and x={5,10}
 are evaluated. Predictor groups are asked to submit a list of predicted contacts and a confidence level for each prediction. The assessors then rank the predictions for each protein and take a look at the top L/x ones, where L is the length of the protein and x={5,10}")
79
Contact Map prediction in CASP
From these L/x top ranked contacts two measures are computed Accuracy: TP/(TP+FP) Xd: difference between the distribution of predicted distances and a random distribution
 Xd: difference between the distribution of predicted distances and a random distribution.")
80
CASP9 results These two groups derived contact predictions from 3D models
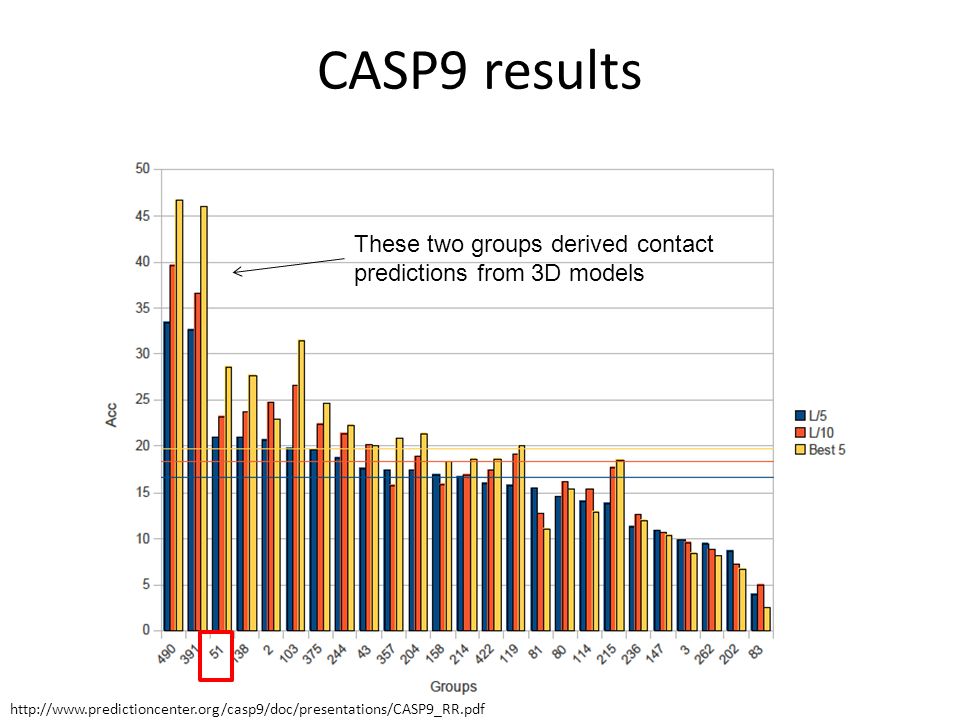
81
Other CASP prediction categories
Functional sites prediction Predicting which residues of a given sequence are those that perform the chemistry of the protein Bind to other proteins/compounds Methods can use whatever information they can infer to perform this prediction However, most predictions can be performed simply by homology Domain prediction Domains = quasi-independent subsets of a protein, that fold on their own Their prediction follows a simple divide-and-conquer motivation It is much easier to create separate models for the different domains of a protein

82
Disordered regions prediction
Regions of a protein that do not fold into a unique pattern (no coordinates in the PDB file) 75% of mammal signaling proteins are estimated to contain long (>30) disordered regions, and 25% of the total amount of proteins may be fully disordered Thus, it is useful to predict from the sequence if that is the case
 75% of mammal signaling proteins are estimated to contain long (>30) disordered regions, and 25% of the total amount of proteins may be fully disordered. Thus, it is useful to predict from the sequence if that is the case.")
83
Disordered protein 2K5K

84
Quality assessment prediction
Given a model, can we predict how good it is (without comparing it to the native structure)? Overall and per-residue model quality Prediction was done based on the models from the server category Two families of methods That perform predictions for individual models That take a set of models and give predictions based on consensus agreements
 Overall and per-residue model quality. Prediction was done based on the models from the server category. Two families of methods. That perform predictions for individual models. That take a set of models and give predictions based on consensus agreements.")
85
Summary of topic Importance of PSP
Many different types of prediction included in the PSP family 3D PSP Prediction of amino acid structural features Others Families of 3D PSP Template-based Modelling Free modelling

Similar presentations











 Protein Structure Prediction Protein Secondary Structure.>")

 Dr. Ahmed Mujamammi Dr. Sumbul Fatma>")





