Download presentation
Presentation is loading. Please wait.

1
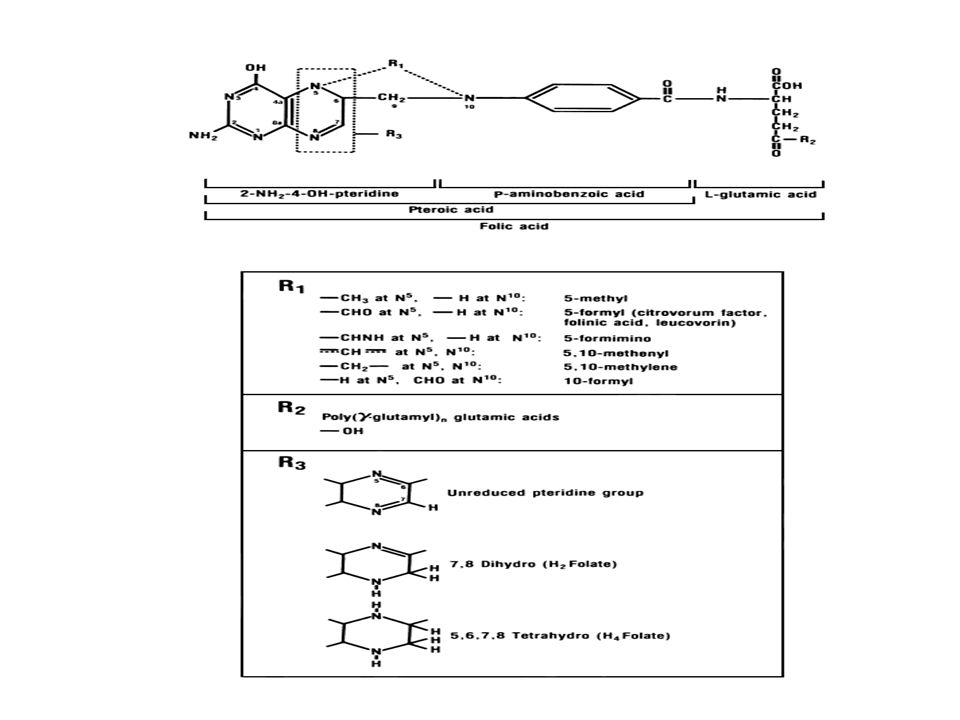
STRUCTURE OF TETRAHYDROFOLATE

3
STRUCTURE OF FOLIC ACID AND REDUCED FOLATES INVOLVED IN ONE-CARBON METABOLISM

5
Inborn Errors of Folate Transport and Metabolism
Hereditary Folate Malabsorption Glutamate Formiminotransferase Deficiency Methylenetetrahydrofolate Reductase Deficiency Methionine Synthase Reductase Deficiency (cblE) Methionine Syntase Deficiency (cblG)
 Methionine Syntase Deficiency (cblG)")
6
HERDITARY FOLATE MALABSORPTION

7
Hereditary Folate Malabsorption
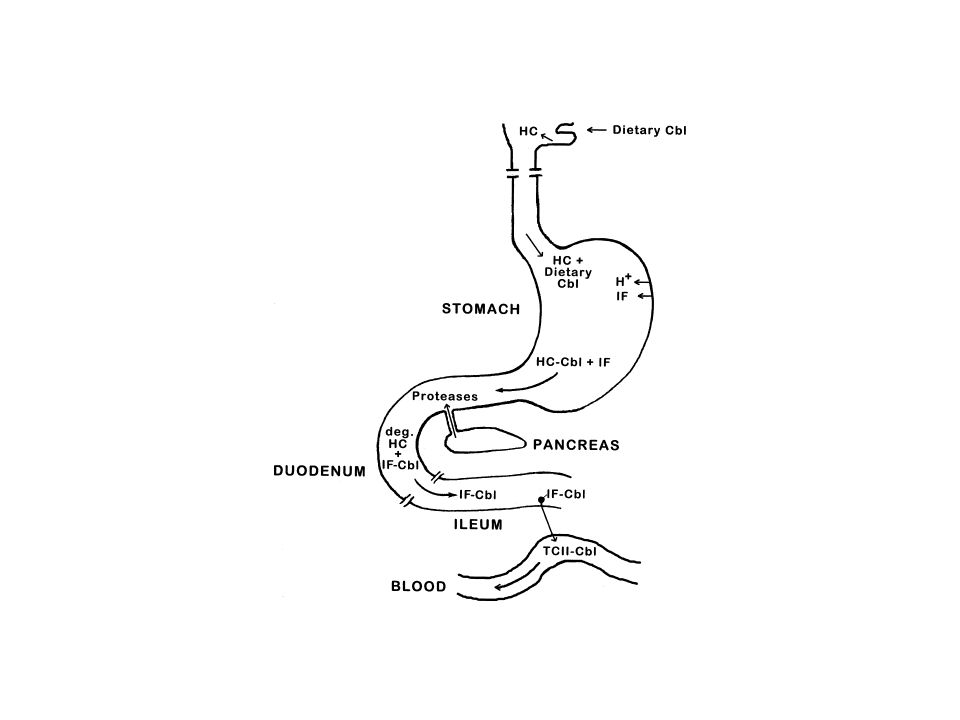
Hereditary folate malabsorption (HFM) (OMIM ) is a rare autosomal recessive disorder caused by impaired intestinal folate absorption with folate deficiency characterized by anemia, hypoimmunoglobulinemia with recurrent infections, such as Pneumocystis carinii pneumonitis, and recurrent or chronic diarrhea. In many patients, neurological abnormalities such as seizures or mental retardation emerge at some point in early childhood, attributed to impaired transport of folates into the central nervous system 1. When this disorder is diagnosed early, signs and symptoms of HFM can be obviated by parental administration of folates or with higher doses of folates by the oral route 1, 2. If untreated, the disease is fatal and, if treatment is delayed, the neurological deficits can become permanent
 (OMIM ) is a rare autosomal recessive disorder caused by impaired intestinal folate absorption with folate deficiency characterized by anemia, hypoimmunoglobulinemia with recurrent infections, such as Pneumocystis carinii pneumonitis, and recurrent or chronic diarrhea. In many patients, neurological abnormalities such as seizures or mental retardation emerge at some point in early childhood, attributed to impaired transport of folates into the central nervous system 1. When this disorder is diagnosed early, signs and symptoms of HFM can be obviated by parental administration of folates or with higher doses of folates by the oral route 1, 2. If untreated, the disease is fatal and, if treatment is delayed, the neurological deficits can become permanent.")
8
Hereditary Folate Malabsorption
Qui A et al. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell 127, , December 1, 2006 Proton coupled, high affinity folate transporter operating at low pH. Loss of function mutations in HFM PCFT/HCP1

9
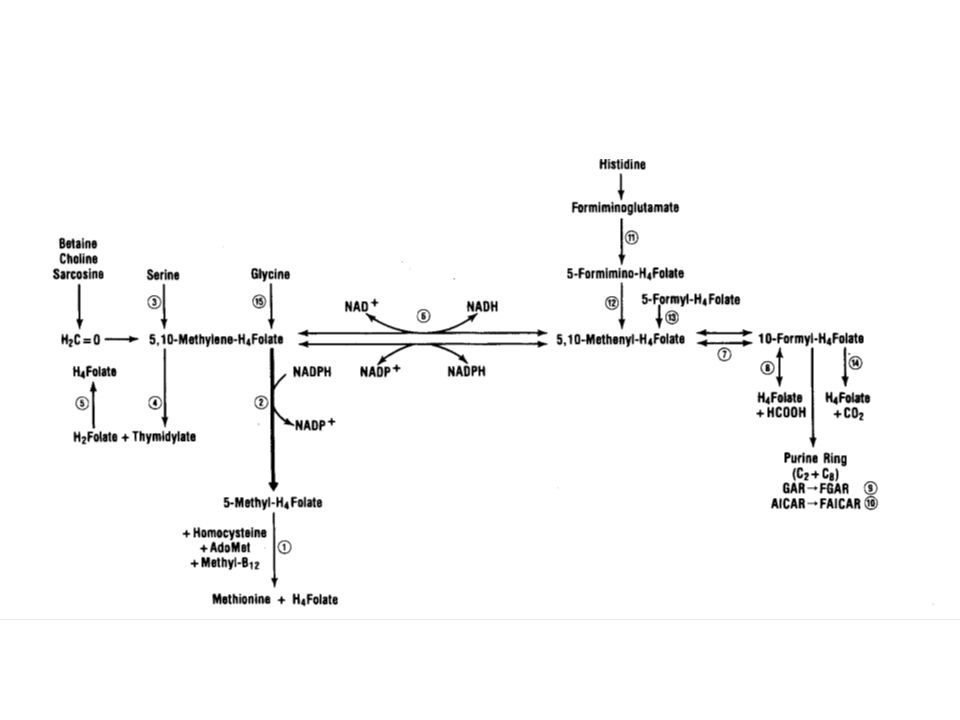
FOLATE PATHWAY

11
Glutamate formiminotransferase
Histidine Formiminoglutamate 2 Glutamate formiminotransferase 5-Formimino-THF Formate + THF 2 Cyclodeaminase 5-Formyl-THF NAD NADH 5, 10-Methenyl-THF 10-Formyl-THF NADP+ NADPH NADP NADPH Purine nucleotides 5, 10-Methylene-THF Methylene-THF reductase dUMP 3 Glycine dTMP 5-Methyl-THF DHF Serine 1 Pyrimidine nucleotides Transport across intestine + CP THF NADPH SAM 4 5 MeCbl Methionine synthase Homocysteine Methionine + THF Figure 1: Summary of major reactions of folate pathway. DHF= dihydrofolate, THF= tetrahydrofolate, dUM= deoxy-uridine phosphate, dTMP= deoxy-thymidine phosphate, CP= choroid plexus, SAM= S-adenosylmethionine, MeCbl= methylcobalamin. Disorders are indicated by circled numbers. 1= Hereditary folate malabsorption, 2= Glutamate formiminotransferase-cyclodeaminase deficiency, 3= Severe Methylenetetrahydrofolate reductase deficiency, 4= Methionine synthase deficiency (cblG) (see Intracellular Cobalamin Metabolism section), 5= Methionine synthase reductase deficiency (cblE) (see Intracellular Cobalamin Metabolism section).
 (see Intracellular Cobalamin Metabolism section), 5= Methionine synthase reductase deficiency (cblE) (see Intracellular Cobalamin Metabolism section).")
12
SEVERE METHYLENETE-TRAHYDROFOLATE (MTHFR) REDUCTASE DEFICIENCY
 REDUCTASE DEFICIENCY")
13
Methylenetetrahydrofolate Reductase Deficiency (Severe)
Hyperhomocysteinemia and homocystinuria Low or normal plasma methionine No megaloblastic anemia !! Variable clinical manifestations including: 1) death in the first year of life; 2) developmental delay; 3) neurologic and psychiatric disease; 4) thrombotic events; 5) asymptomatic Gene/location: MTHFR/ Chr. 1p36.3 Common polymorphisms: 677CT; 1298AC
 death in the first year of life; 2) developmental delay; 3) neurologic and psychiatric disease; 4) thrombotic events; 5) asymptomatic. Gene/location: MTHFR/ Chr. 1p36.3. Common polymorphisms: 677CT; 1298AC.")
15
COMMON POLYMORPHISMS IN MTHFR

16
MTHFR 677CT Originally discovered because specific activity of MTHFR in cell extracts was thermolabile 50-60% decrease in specific activity of MTHFR First postulated association (Kang et al) was between thermolability of MTHFR and heart disease
 was between thermolability of MTHFR and heart disease.")
17
MTHFR 677CT After cloning of the gene, the cause of thermolability of MTHFR was shown to be this common polymorphism in the catalytic domain that results in the change of an alanine to a valine. Gene frequency of the T allele varies with ethnic groups (30% in Europeans and Japanese, 11% in African Americans).
.")
18
MTHFR 677CT T allele is associated with elevated levels of total homocysteine (tHcy). Effect is much more prominent in TT individuals Dietary folate (multivitamins, fortification of cereal grains) can mask the effect of the T allele.
 can mask the effect of the T allele.")
19
MTHFR 677CT Disease Associations (Incomplete)
Cardiovascular Disease Alzheimer Disease Colon Cancer Diabetes Mellitus Down Syndrome Leukemia Neural Tube Defects (NTD) Pregnancy Complications
 Pregnancy Complications.")
20
MTHFR 1298AC Associated with 35% decrease in MTHFR specific activity
Not associated with enzyme thermolability Frequency of C allele: 30% Western Europe and 18% in Asians 1298C and 677T rarely found together in cis Fewer studies have looked at this polymorphism

21
GLUTAMATE FORMININOTRANSFERASE DEFICIENCY

23
Glutamate Formimotransferase Deficiency
Autosomal Recessive (<20 patients) Formiminoglutamate (FIGLU) excretion Clinical heterogeneity: 1) developmental delay, elevated serum folate, FIGLU excretion 2) mild speech delay, high levels of FIGLU excretion. Note that GFTD activity cannot be measured in cultured cells-present only in liver.
 Formiminoglutamate (FIGLU) excretion. Clinical heterogeneity: 1) developmental delay, elevated serum folate, FIGLU excretion 2) mild speech delay, high levels of FIGLU excretion. Note that GFTD activity cannot be measured in cultured cells-present only in liver.")
26
Human FTCD Discovered by examination of EST’s on chromosome 21 as part of a study assessing the molecular basis of Down Syndrome EST compared to porcine FTCD Human 21q22.3 15 exons 541 amino acid residues with 84% homology to the pig. Five different transcripts

28
GFT Patients Siblings: 1) Age 2 1/2 years - speech delay, some growth delay, hypotonia, increased FIGLU excretion 2) Age 8 years-hypotonia, abnormal EEG, increased FIGLU excretion Two missense mutations: c457 c->T (R135C) and c940 C->G (R299P). Not found in 200 control alleles.
 Age 2 1/2 years - speech delay, some growth delay, hypotonia, increased FIGLU excretion 2) Age 8 years-hypotonia, abnormal EEG, increased FIGLU excretion. Two missense mutations: c457 c->T (R135C) and c940 C->G (R299P). Not found in 200 control alleles.")
31
Third GFT Patient Apnea in the first year of life Recurrent infections
At age 2, mild developmental delay, hypotonia, breathing difficulties Hypersegmented neutrophils Increased FIGLU excretion One mutation: c1033 insG (not found in 200 control alleles)
")
33
Southern Blot HindIII BamHI Kpn I MCH24 WG1795 MCH39 WG1191 MCH24
10 ug of genomic DNA (5 ug for MCH 39) was digested with the indicated enzymes, run on a 0.8% agarose gel at 25V and transferred to Hybond N+. The blot was probed with random-primed P32 labelled hFTCD (B-form) probe.
 was digested with the indicated enzymes, run on a 0.8% agarose gel at 25V and transferred to Hybond N+. The blot was probed with random-primed P32 labelled hFTCD (B-form) probe.")
34
Western Blot c1033insG FTCDH6 CD333H6 S407L FTH6 R135C R299P A438E
175 kDa 83.0 kDa 62.0 kDa 47.5 kDa 32.5 kDa 25.0 kDa 16.5 kDa 25 ? Ug of protein (crude extract) was run on 12%SDS-PAGE and transferred to nitrocelluose. The blot was probed with polyclonal rabbit anti-pFTCD followed by HRP-conjugated goat anti-rabbit IgG.
 was run on 12%SDS-PAGE and transferred to nitrocelluose. The blot was probed with polyclonal rabbit anti-pFTCD followed by HRP-conjugated goat anti-rabbit IgG.")
37
FTCD Assay

38
FTCD Assay

39
Conclusions First mutations in Human FTCD in three patients with glutamate formiminotransferase deficiency.

40
FUNCTIONAL METHIONINE SYNTASE DEFICIENCY
Overlap in Folate and Cobalamin Metabolism: One phenotype Two Genotypes: cblE (Methionine synthase reductase deficiency) cblG (Methionine synthase deficiency)
 cblG (Methionine synthase deficiency)")
42
METHIONINE SYNTHASE REDUCTASE DEFICIENCY (cblE)
")
43
Methionine Synthase Reductase Deficiency-cblE
Megaloblastic anemia, hyperhomocysteinemia and homocystinuria Low plasma methionine Cerebral atrophy, nystagmus, blindness, altered tone Reduced methionine synthase activity in the absence of an exogenous reducing system Gene/ location: MTRR/ 5p Polymorphism: 66AG

44
Methylcobalamin-Dependent Methionine Synthase in E. Coli
2 component flavoprotein system flavodoxin NADPH-ferredoxin (flavodoxin) oxidoreductase, a member of electron transferases termed the “FNR family”
 oxidoreductase, a member of electron transferases termed the FNR family")
45
Methionine Synthase Reductase
Findings suggest evolution of the two genes specifying flavodoxin/flavodoxin reductase to a single gene encoding a fused version of the two proteins in man. This new gene has been called MTRR since the gene for methionine synthase is MTR.

46
Methionine Synthase Reductase
Localized to chromosome 5p15.2-p15.3 2094 bp amino acids Predicted molecular mass 77,000 Da Prominent RNA species of 3.6 kb with an additional smaller 3.1 kb species in brain 38% identity (49% similarity) with human cytochrome P-450 reductase
 with human cytochrome P-450 reductase.")
47
Lysosome Mitochondrion Methylmalonyl-CoA TCII- Cob(III)alamin mut cblB TCII Cob(I)alamin AdoCbl Methylmalonyl-CoA Mutase Cob(III)alamin cblF cblA Succinyl-CoA cblH Cob(III)alamin Cob(II)alamin cblC cblD Cob(I)alamin 5-MethylTHF Methionine MTHFR Methionine Synthase cblG Cob(II)alamin cblG 5,10-methyleneTHF Methionine Synthase Reductase AdoMet Extracellular Space Cytoplasm THF cblE Homocysteine Methylcobalamin
alamin. cblF. cblA. Succinyl-CoA. cblH. Cob(III)alamin. Cob(II)alamin. cblC. cblD. Cob(I)alamin. 5-MethylTHF. Methionine. MTHFR. Methionine Synthase. cblG. Cob(II)alamin. cblG. 5,10-methyleneTHF. Methionine Synthase Reductase. AdoMet. Extracellular Space. Cytoplasm. THF. cblE. Homocysteine. Methylcobalamin.")
49
METHIONINE SYNTHASE DEFICIENCY (cblG)
")
50
Methionine Synthase Deficiency-cblG
Hyperhomocysteinemia and homocystinuria Low plasma methionine; Megaloblastic anemia Cerebral atrophy, nystagmus, blindness, altered tone. Some patients present in adult life!! Reduced methionine synthase activity Gene/Location: MTR/ Chr. 1q43 Polymorphism: 2756AG

53
Table 155-2: Inherited Defects of Folate Metabolism

56
:

57
I-F-Cobalamin Receptor Deficiency
(Imerslund –Gräsbeck Syndrome) (MGA1) Example of One Phenotype, 2 Genes
 (MGA1) Example of One Phenotype, 2 Genes.")
58
I-F-Cobalamin Receptor Deficiency (Imerslund -Gräsbeck) (MGA1)
Early onset megaloblastic anemia, low serum cobalamin levels, and proteinuria Homocystinuria and methylmalonic aciduria may be found but are not prominent Decreased absorption of cobalamin in the presence of normal synthesis of intrinsic factor Common in Finland, Norway and the Middle East Defects in CUBN (cubilin) & AMN (amnionless) Genes/ Locations: Chrs. 10p12.1 & 14q32
 & AMN (amnionless) Genes/ Locations: Chrs. 10p12.1 & 14q32.")
59
I-F-Cobalamin Receptor Deficiency (Imerslund -Gräsbeck) (MGA1)
Fyfe et al. Blood Online October 2003: Interaction of cubilin and amnionless to form a complex (cubam) that functions as the cobalamin-IF receptor. Without amnionless, cubilin does not reach the cell membrane.
 that functions as the cobalamin-IF receptor. Without amnionless, cubilin does not reach the cell membrane.")
60
Intracellular Cobalamin Metabolism:
Endocytosis Reduction Mitochondrial Transport & Adenosylation-AdoCbl Methylation-MeCbl

61
Lysosome Mitochondrion Methylmalonyl-CoA TCII- Cob(III)alamin mut cblB TCII Cob(I)alamin AdoCbl Methylmalonyl-CoA Mutase Cob(III)alamin cblF cblA Succinyl-CoA cblH Cob(III)alamin Cob(II)alamin cblC cblD Cob(I)alamin 5-MethylTHF Methionine MTHFR Methionine Synthase cblG Cob(II)alamin cblG 5,10-methyleneTHF Methionine Synthase Reductase AdoMet Extracellular Space Cytoplasm THF cblE Homocysteine Methylcobalamin
alamin. cblF. cblA. Succinyl-CoA. cblH. Cob(III)alamin. Cob(II)alamin. cblC. cblD. Cob(I)alamin. 5-MethylTHF. Methionine. MTHFR. Methionine Synthase. cblG. Cob(II)alamin. cblG. 5,10-methyleneTHF. Methionine Synthase Reductase. AdoMet. Extracellular Space. Cytoplasm. THF. cblE. Homocysteine. Methylcobalamin.")
62
MMA Is the MMA isolated? Is tHcy elevated?
Low serum cobalamin levels should lead one to expect a disorder of intake or transport: Breast –fed infant of vegan mother or mother with subclinical PA Imersund-Grasbeck (MGA1)-mutations in cublin or amnionless (Stephan Tanner-Ohio) Combined MMA and Homocystinuria (cblC, cblD, cblF)
-mutations in cublin or amnionless (Stephan Tanner-Ohio) Combined MMA and Homocystinuria (cblC, cblD, cblF)")
63
MUT

64
mut MMA At least 178 different mutations
Difficult to make genotype/phenotype correlations. Many patients are compound heterozygotes and different patients homozygous for the same mutation may have different phenotypes There are a number of mutations that are more common in specific ethnic groups and a number of common mutations.

65
MUT Missense Mutations Nonsense Mutations Deletions and insertions
seen in more than one patient seen in only one family Missense Mutations Nonsense Mutations Deletions and insertions Splice Mutations

66
Cobalamin-responsive MMA
Two genes cloned on the basis of homology: MMAA: cblA complementation group MMAB: cblB complementation group

68
MMAA

69
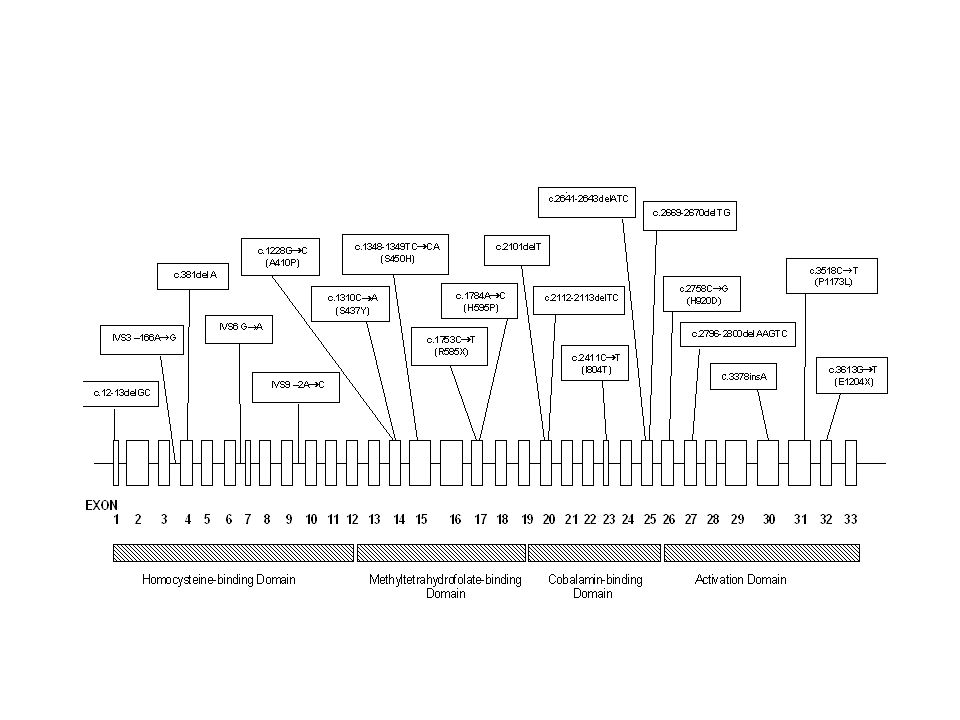
Exon c.64C>T (R22X) c.161G>A (W54X) c.260-267dupATAAACTT
c.266T>C (L89P ) c.283C>T (Q95X) c.387C>A (Y129X) c.742C>T (Q248X) c.433C>T (R145X) c.959G>A (W320X) c.434G>A (R145Q) c.439+1_4delGTCA Splice c.970-2A>T Splice Exon 1 2 3 4 5 6 7 c.733+1G>A Splice c.620A>G (Y207C) c.653G>A (G218E) c.592_595delACTG c.988C>T (R330X) c.503delC c.1076G>A (R359Q) c.450_451insG c.440G>A (E147G) c.1089_1090delGA
 c.283C>T (Q95X) c.387C>A (Y129X) c.742C>T (Q248X) c.433C>T (R145X) c.959G>A (W320X) c.434G>A (R145Q) c.439+1_4delGTCA Splice. c.970-2A>T Splice. Exon c.733+1G>A Splice. c.620A>G (Y207C) c.653G>A (G218E) c.592_595delACTG. c.988C>T (R330X) c.503delC. c.1076G>A (R359Q) c.450_451insG. c.440G>A (E147G) c.1089_1090delGA.")
70
MMAA At least 29 mutations known
C.433C>T accounts for 43% alleles in one North American Study c503delC more frequent in Japan (8 of 14 mutant alleles)
")
71
MMAB

72
MMAB c.572_576 del GGGCC c.56-57 GC>AA (R19Q) c.575 G>A (E193K)
c.716 T>A (M239K) c.IVS2-1 G>T c.571 C>T (R191W) c.700 C>T (Q234X) c.569 G>A (R190H) c.656 A>G (Y219C) c.IVS7-2 A>C c.556 C>T (R186W) c.654_657 del CTAT c.403 G>A (A135T) c.IVS3-1 G>A
 c.IVS2-1 G>T. c.571 C>T (R191W) c.700 C>T (Q234X) c.569 G>A (R190H) c.656 A>G (Y219C) c.IVS7-2 A>C. c.556 C>T (R186W) c.654_657 del CTAT. c.403 G>A (A135T) c.IVS3-1 G>A.")
73
MMAB Mutations 22 mutations Identified
Most predicted to affect the active site of the enzyme, identified from the crystal structure of is bacterial ortholog C.556C>T (p.R186W) represents 33% of affected alleles.
 represents 33% of affected alleles.")
74
MMADHC

75
MMADHC-cblD variant=cblH
Associated with isolated MMA Decreased propionate incorporation Decreased AdoCbl synthesis Novel gene MMADHC isolated by Brian Fowler in Switzerland Identical to cblH Mutations in N-terminal regions associated with isolated MMA

76
cblD-HC cblD-MMA cblD-HC+MMA 2 4 5 6 7 8 3 L20fsX21 S228M T152fsX162 9
478 609 696 891 154 372 cblD-MMA cblD-HC+MMA cblD-HC L20fsX21 S228M T152fsX162

77
Genes Associated with Isolated MMA
MUT MMAA MMAB MMADHC (NEJM in press) MCEE-may not be related to clinical SUCLA2-developmental delay SUCLG1-fatal infantile lactic acidosis (Ostergaard E et al. Am J Hum Genet 81:383, 2007)
 MCEE-may not be related to clinical. SUCLA2-developmental delay. SUCLG1-fatal infantile lactic acidosis (Ostergaard E et al. Am J Hum Genet 81:383, 2007)")
78
Methylmalonyl-CoA Succinyl-CoA
5-Methyl-THF THF cblG Methionine synthase Homocysteine Methionine MeCbl MTRR cblE Co(I)bl cblD variant 1 cblC, cblD cblF Cbl TC TC/Cbl Co(III)bl Co(II)bl cblD variant 2 Lysosome cblA, cblH Co(II)bl Co(I)bl cblB AdoCbl Cell membrane Methylmalonyl-CoA Succinyl-CoA Methylmalonyl-CoA mutase mut Mitochondrion
bl. cblD variant 1. cblC, cblD. cblF. Cbl. TC. TC/Cbl. Co(III)bl. Co(II)bl. cblD variant 2. Lysosome. cblA, cblH. Co(II)bl. Co(I)bl. cblB. AdoCbl. Cell membrane. Methylmalonyl-CoA Succinyl-CoA. Methylmalonyl-CoA. mutase. mut. Mitochondrion.")
79
cblC Most common inborn error of Vitamin B12 metabolism Early-onset:
Feeding difficulties, hypotonia/hypertonia, lethargy Abnormal movement, seizures Multisystemic involvement Pancytopenia or megaloblastic anemia Salt-and-pepper retinopathy Moderate to severe cognitive disability

80
cblC Late-onset (renal phenotype):
Chronic thrombotic microangiopathic syndrome Absence of neurological involvement Late-onset (neurological phenotype): Sudden cognitive decline (confusion, dementia) Extrapyramidal signs, ataxia, peripheral neuropathy Milder hematological abnormalites
: Sudden cognitive decline (confusion, dementia) Extrapyramidal signs, ataxia, peripheral neuropathy. Milder hematological abnormalites.")
81
Diagnosis of cblC Clinical history, physical exam
Laboratory investigations: CBC with smear, ± bone marrow biospy Plasma amino acids (elevated Hcy, low methionine) Urine organic acids (elevated MMA) Total plasma homocysteine Others as clinically indicated (Normal serum cobalamin and folate levels).
 Urine organic acids (elevated MMA) Total plasma homocysteine. Others as clinically indicated (Normal serum cobalamin and folate levels).")
82
Diagnosis of cblC Special investigations: cultured fibroblasts
Incorporation of label from [14C]propionate and [14C]methyltetrahydrofolate into cellular macromolecules Cbl distribution studies Complementation studies

85
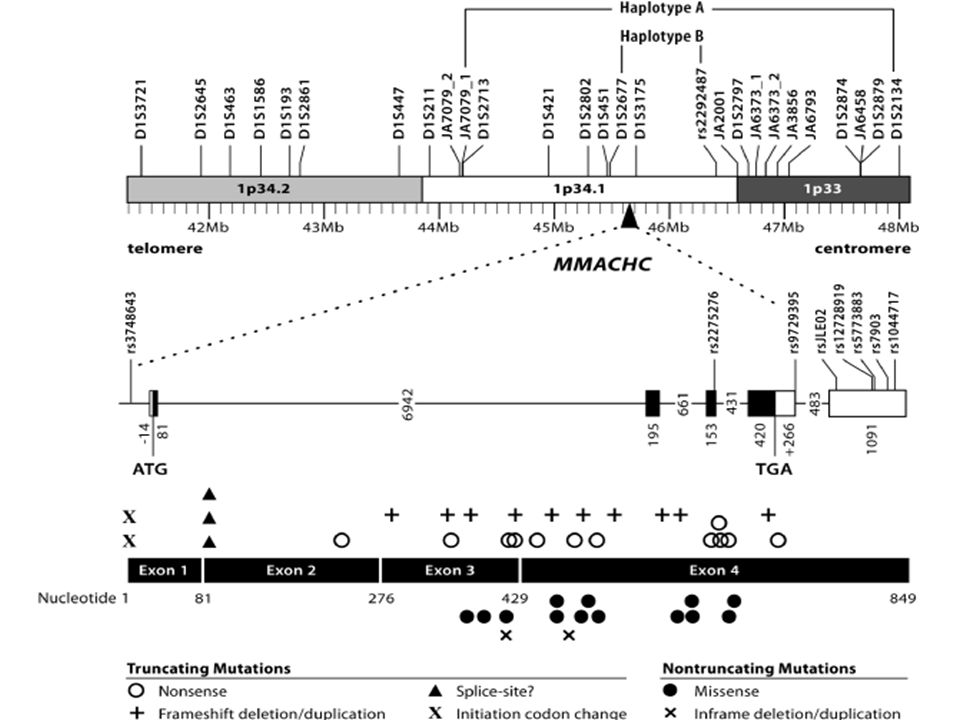
c.331C>T c.440G>A c.271dupA c.3G>A c.394C>T c.608G>A c.547_548delGT c.609G>A

86
Homozygosity mapping and haplotype analysis
MMACHC Some degree of homology with TonB, a bacterial protein involved in energy transduction for vitamin B12-uptake

87
204 patients 42 mutations c.271dupA: 40% c.331C>T: 9% c.394C>T: 8%

88
Phenotype-Genotype Correlations: Seeking Answers in Case-Reports
37 previously published patients: 25 early-onset cases 12 late-onset cases: 9: neurological phenotype 3: renal phenotype

89
Early-Onset Cases 25 out of 37 patients 9/25: homozygous for c.271dupA
3/25: homozygous for c.331C>T 5/25: c.271dupA / c.331C>T 1/25: c.271dupA / c.394C>T Remaining 8 patients either: Compound heterozygous for different nonsense mutations Homozygous for another nonsense mutation

90
Late-Onset Cases 12 of 37 patients 9/12: neurological phenotype
3/12: renal phenotype Neurological phenotype: 4/9: homozygous for c.394C>T 2/9: c.271dupA and c.394C>T 3/9: c.271dupA and a missense mutation Renal phenotype: 3/3: c.271dupA and c.82-9_-12delTTTC

91
Observations on Ethnic Background
Homozygosity for c.271dupA: 9 patients 5 White 1 Hispanic 1 Iranian 1 Middle Eastern 1 ? Ethnicity In database: 44 other patients of various ethnic backgrounds Therefore, not specific to one ethnic group

92
Observations on Ethnic Background
Homozygosity for c.331C>T: 3 patients “Cajun” 3 unpublished French Canadian patients from Québec and New Brunswick Compound heterozygosity c.331C>T/c.271dupA: 5 patients: 1: White (USA, “French” background on pedigree in lab) 1: French Canadian from Québec 3: Louisiana, USA (New Orleans) In database: 5 additional patients of French-Canadian or Cajun background Suggest possible founder effect/genetic drift
 1: French Canadian from Québec. 3: Louisiana, USA (New Orleans) In database: 5 additional patients of French-Canadian or Cajun background. Suggest possible founder effect/genetic drift.")
93
Observations on Ethnic Background
Homozygosity for c.394C>T: 4 patients 3: Asiatic-Indian (incl. 2 sibs) 1: Middle Eastern In database: 9 other patients, all Asiatic-Indian, Pakistani or Middle Eastern Heterozygosity c.394C>T / c.271dupA: 3 patients 1: Greek 1: Portuguese 1: ? Ethnicity Mutational hot-spot: arose at least twice independently
 1: Middle Eastern. In database: 9 other patients, all Asiatic-Indian, Pakistani or Middle Eastern. Heterozygosity c.394C>T / c.271dupA: 3 patients. 1: Greek. 1: Portuguese. 1: Ethnicity. Mutational hot-spot: arose at least twice independently.")
94
Observations on Ethnic Background
Homozygosity for c.440G>A: 2 patients Native American (Southwestern) In database: 1 unpublished Native American patient of the same tribe
 In database: 1 unpublished Native American patient of the same tribe.")
95
Compound heterozygosity: c.394C>T / c.271dupA
2 late-onset published cases: Ages 4.5 and 10 years 1 early-onset published case: Age 6 months Intrafamilial phenotypic heterogeneity: Augoustides-Savvopoulou P, Mylonas I, Sewell AC, Rosenblatt DS. Reversible dementia in an adolescent with cblC disease: clinical heterogeneity within the same family. J InheritMetab Dis 1999; 22(6): Late-onset AND early-onset in the same family!!! Interpretation of anticipated phenotype based on this genotype may be unreliable
: Late-onset AND early-onset in the same family!!! Interpretation of anticipated phenotype based on this genotype may be unreliable.")
96
Response to Cbl Supplementation
Homozygosity c.271dupA: Tend to have progression of disease despite Tx Homozygosity c.394C>T: Almost complete reversal of psychiatric and neurological symptoms Compound heterozygosity c.394C>T / c.271dupA

97
c.271dupA / missense mutations
Late-onset neurological phenotype: c.271dupA / c.440G>C, 45 years Powers JM, Rosenblatt DS, Schmidt RE et al. Neurological and neuropathologic heterogeneity in two brothers with cobalamin C deficiency. Ann Neurol 2001; 49(3): c.271dupA / c.482G>A, 20 years Bodamer OA, Rosenblatt DS, Appel SH, Beaudet AL. Adult-onset combined methylmalonic aciduria and homocystinuria (cblC). Neurology 2001; 56(8):1113 c.271dupA / c.347T>C, 24 years Roze E, Gervais D, Demeret S et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol 2003; 60(10):
: c.271dupA / c.482G>A, 20 years. Bodamer OA, Rosenblatt DS, Appel SH, Beaudet AL. Adult-onset combined methylmalonic aciduria and homocystinuria (cblC). Neurology 2001; 56(8):1113. c.271dupA / c.347T>C, 24 years. Roze E, Gervais D, Demeret S et al. Neuropsychiatric disturbances in presumed late-onset cobalamin C disease. Arch Neurol 2003; 60(10):")
102
The MMACHC protein is not a member of any previously identified gene family.
It is well conserved among mammals. However, the C-terminal end does not appear to be conserved in eukaryotes outside Mammalia, and no homologous protein was identified in prokaryotes Motifs were identified in MMACHC that are homologous to motifs in bacterial genes with vitamin B12-related functions.

105
It is possible that the MMACHC gene product plays a role, directly or indirectly, in removal of the upper axial ligand and/or reduction of Cbl, and this is a challenge for future studies. MMACHC may be involved in the binding and intracellular trafficking of Cbl. Further studies on co-localization and a search for novel binding partners may help us to better understand the early steps of cellular vitamin B12 metabolism.

106
Cobalamin metabolism Moras et al., 2006

107
:

Similar presentations