Download presentation
Presentation is loading. Please wait.

1
Disorder of the sex development Rickets
A. Luczay

2
Sex development GENETIC X X X Y (chromosal) GONADAL ovarium testis
GENITAL INTERNAL uterus prostate EXTERNAL female male SEX ASSIGNMENT girl boy PSYCHOSOCIAL female male

3
DIFFERENTIATION OF THE GONADS
PRIMORDIAL GERM CELS ADRENAL MEDULLA ADRENAL CORTEX WOLFFIAN DUCT MÜLLERIAN DUCT MEDULLA CORTEX BIPOTENTIAL GONAD 46 XX 46 XY DEVELOPING TESTIS DEVELOPING OVARY TUBULUBI SEMINIFERIS WOLFFIAN DEGENERATION CONDUCTING DUCT PRIMARY FOLLICLES SPERMATO- GONIUMS MÜLLERIAN DUCT REGRESSION FALLOPIAN TUBE

4
GENITAL DIFFERENTIATION
INDIFFERENT STAGE GONAD MESONEPHROS MÜLLERIAN DUCT WOLFFIAN DUCT UROGENITAL FOLD INDIFFERENT STAGE LABIOSCROTAL SWELLING MALE FEMALE GLANS UROGENI- TAL FOLD FUSIONED UROGENITAL FOLD URETH-RAL SLIT EPIDIDYMIS OVARY TESTIS FALLO-PIAN TUBE VAS DEFERENS ANUS GLANS PENIS URETHRAL MEATUS UTERUS SEMINAL VESICLE CLITORIS PROSTATE VAGINAL ORIFICE VAGINA RAPHE FEMALE MALE

5
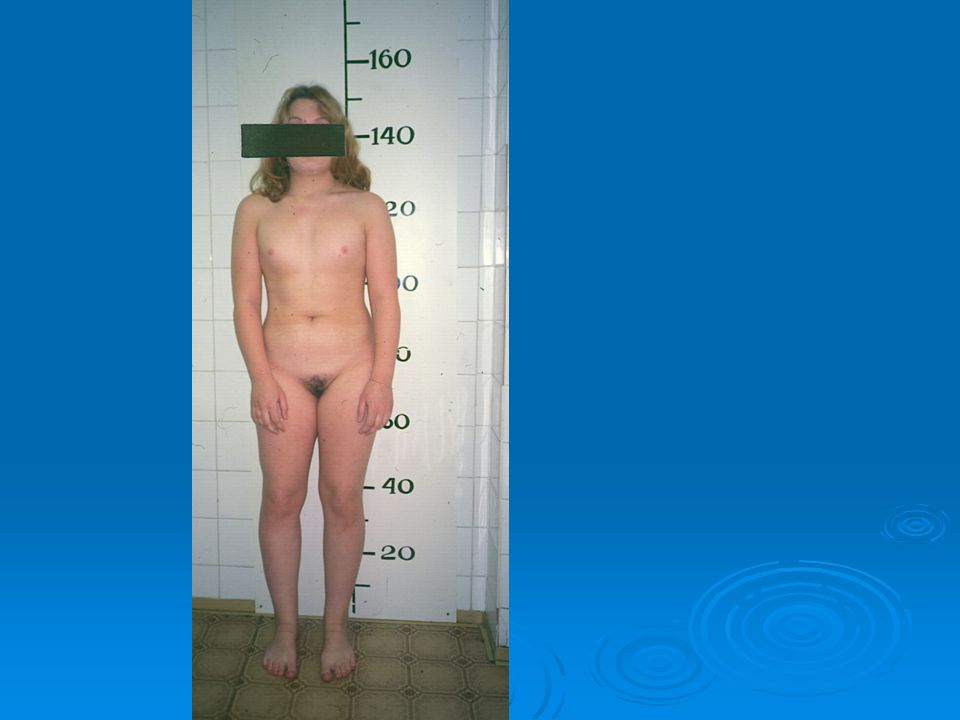
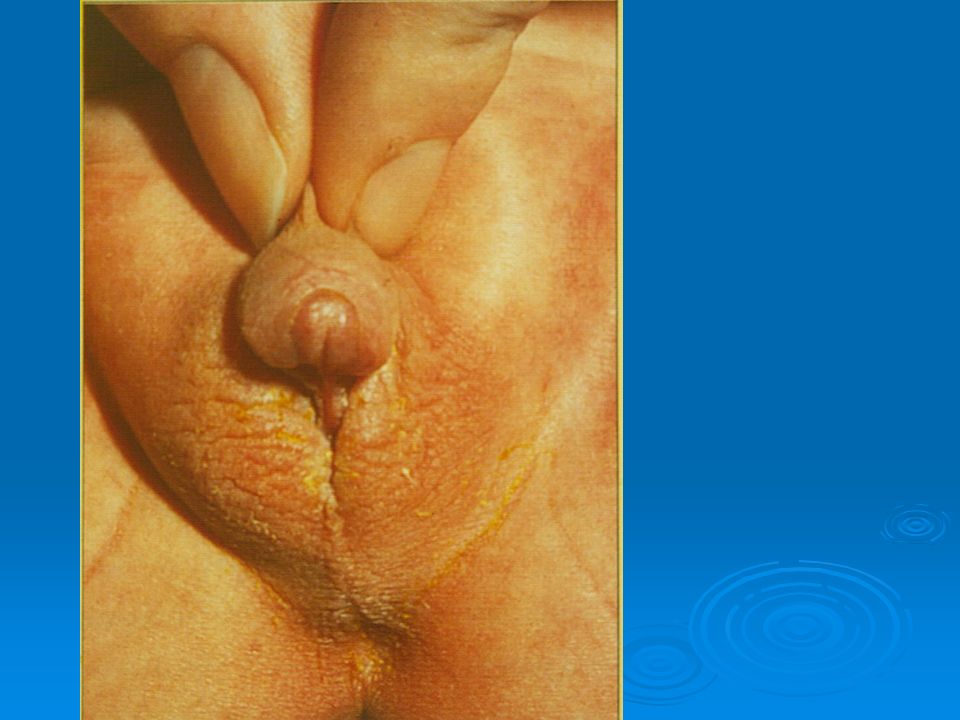
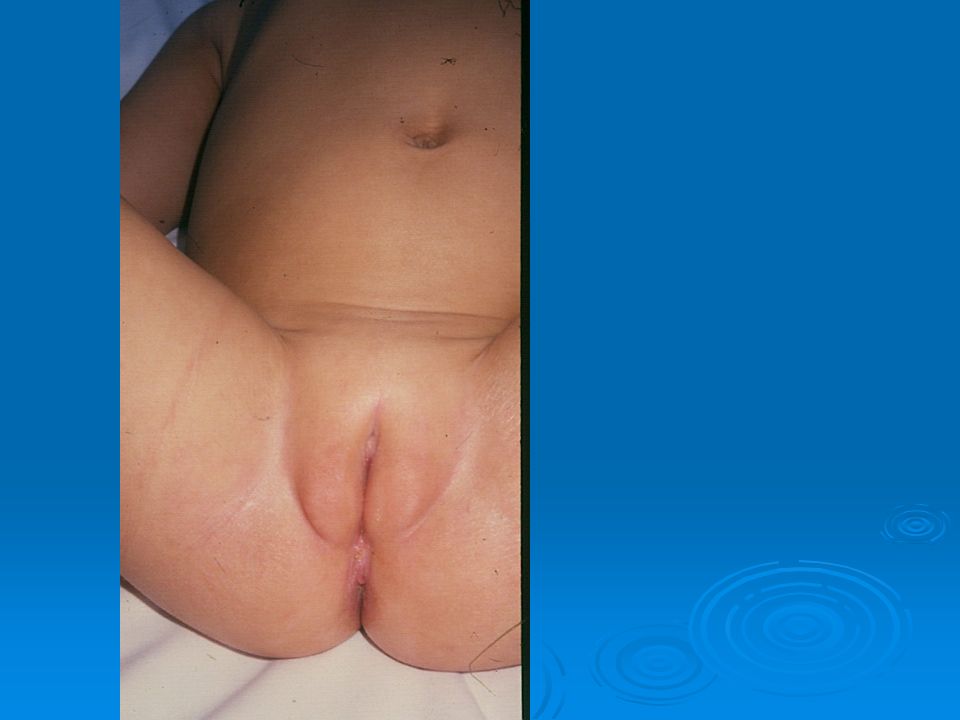
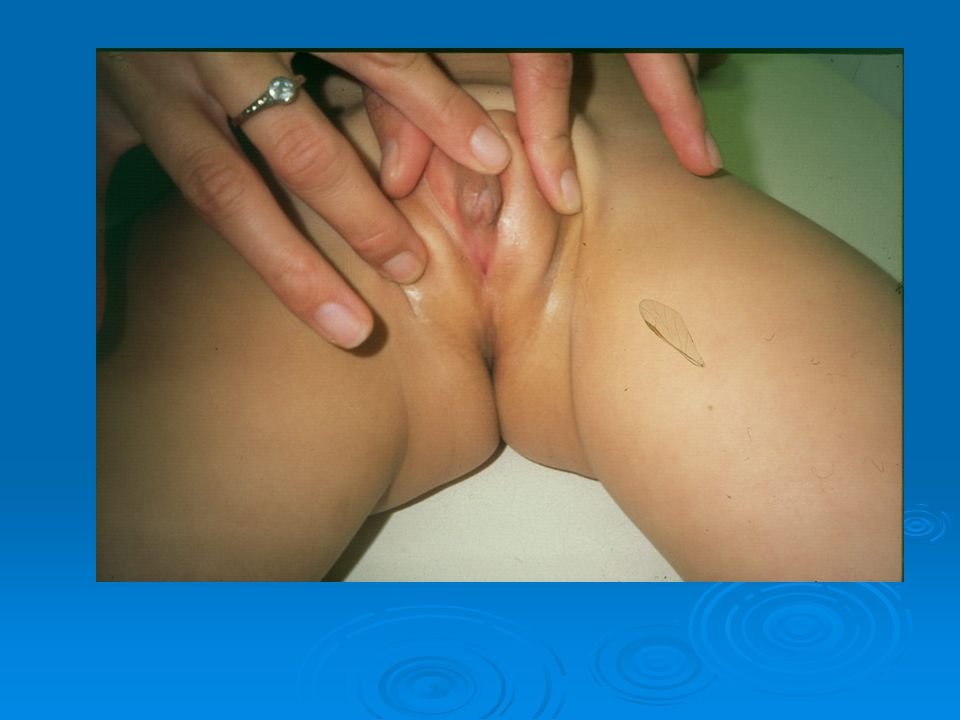
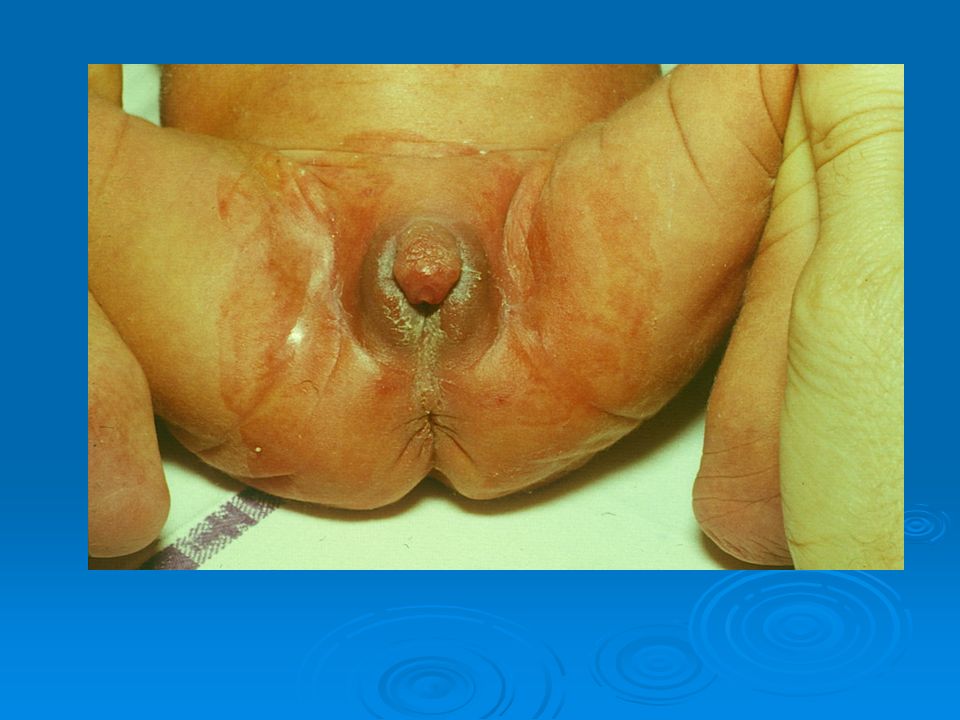
DISORDERS OF THE EXTERNAL GENITALIA
SINECKER PRADER

6
CLINICAL ASSESSMENTOF INFANTS WITH AMBIGUOUS GENITALIA
GONADS PALPABLE NON-PALPABLE NORMAL NORMAL INCREASED KARYOTYPE SERUM Te HIGH LOW

7
Classification

8
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
9
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
10
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
13
Turner syndrome 1/2500 live female birth Hand-food edema in infancy
Pterygium colli (neck webbing), Low posterior hairline, Broad chest, short stature Cardiac, renal malformations Karyotype: 45,X
, Low posterior hairline, Broad chest, short stature. Cardiac, renal malformations. Karyotype: 45,X.")
14
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
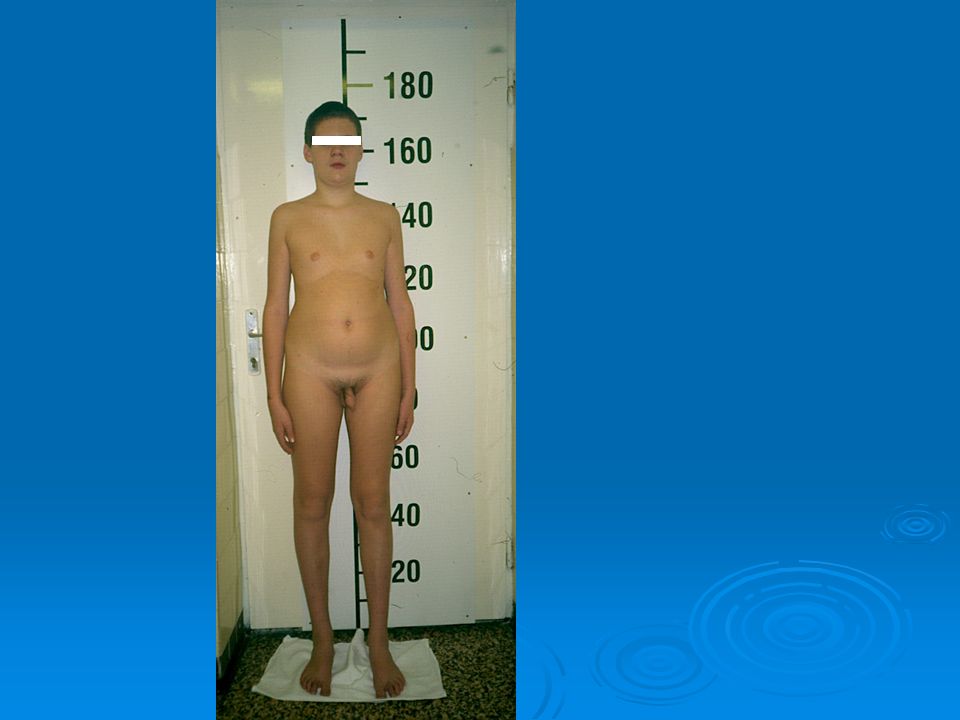
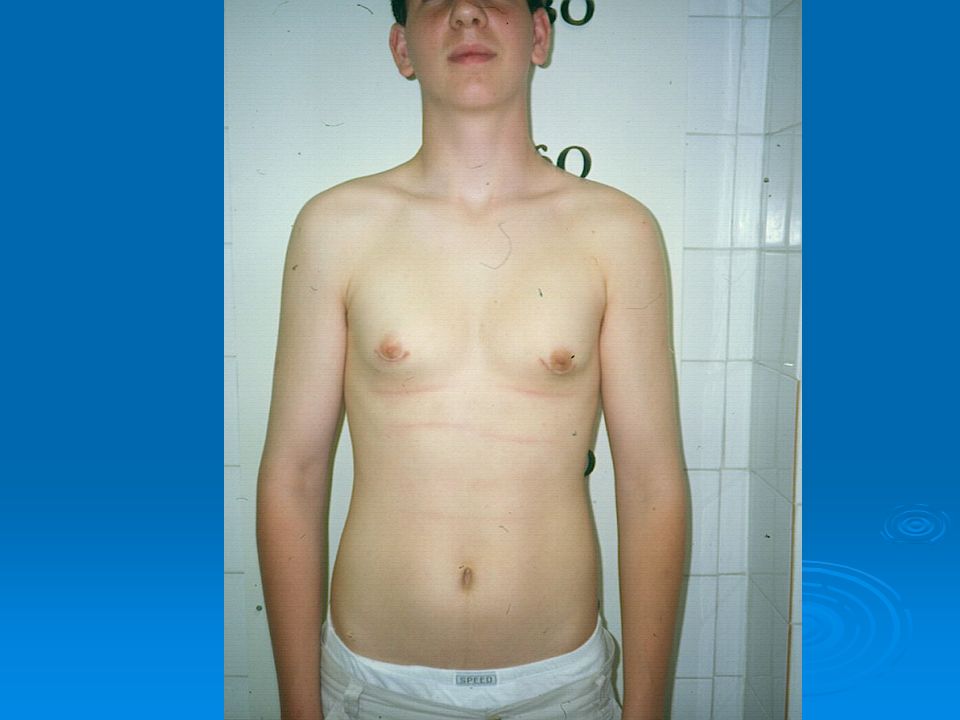
18
Klinefelter syndrome 1/500-1000 live male birth
Small testis, high stature, learning difficulties, gynecomastia in puberty At pubert testicular size increases (~10 ml) Midpuberty: low androgen level Karyotype: 47,XXY
 Midpuberty: low androgen level. Karyotype: 47,XXY.")
19
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
21
Complet gonadal dysgenesis Swyer syndrome
Bilateral streak gonads Apparently normal female external genitalia High risk of gonadoblastoma, germinoma Karyotype: 46,XY

22
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovotesticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
24
Parcial gonadal dysgenesis
Ambigous genitalia (Leydig cell mass) Partial rest of Müllerian duct Karyotype: 46,XY High risk of gonadoblastoma
 Partial rest of Müllerian duct. Karyotype: 46,XY. High risk of gonadoblastoma.")
25
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
27
Steroid Hormone Biosynthesis
dehidroepiandrosterone HO O 17a-OH-progesterone cholesterol ALDOSTERONE Pregnenolone Progesterone 11-Dezoxycortisol Corticosterone CORTIZOL 17a-OH-pregnenolone C CH3 DOC CH2OH OCH OH 17 ANDROSTENEDIONE

28
17- hydroxilase defect Rare form of CAH
Both testosterone and estrogen synthesis is decreased Hypertension, hypokalemia

29
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
31
Complete androgen insensitivity
X q11-12 – AR gene Female external genitalia, good breast development, hairless Low risk of gonadoblstoma (2-5%)
")
32
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
34
Parcial androgen insensitiviy
X q11-12 The seerity of undervirilisation depend on the receptor sensitivity. High risk of gonadoblastoma

35
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen over production 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen over production. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defektus. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
37
Steroid Hormone Biosynthesis
dehidroepiandrosterone HO O 17a-OH-progesterone cholesterol ALDOSTERONE Pregnenolone Progesterone 11-Dezoxycortisol Corticosterone CORTIZOL 17a-OH-pregnenolone C CH3 DOC CH2OH OCH OH 17 ANDROSTENEDIONE P450c21

38
Cngenital adrenal hyperplasia (21-OHD)
1/15000 live birth CYP21 gene mutation 2/3 salt wasting form

39
SEX CHROMOSOME DSD DSD KARYOTYPE : 46,XY DSD KARYOTYPE: 46,XX Disorder of testicular development Disorder of ovarian development 45,X Turner syndrome 1. Complet Gonadal Dysgenesis (Swyer syndroma) 1. Ovotesticular DSD 2. Partial Gonadal Dysgenesis 2. testis SRY+, SOX9 duplication 3. Testicular regression 3. gonadal dysgenesis 4. Ovotesticular DSD 47, XXY Klinefelter syndrome Defect in androgen biosynthesis or in androgen action Androgen overproduction 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defect 3. maternal origin: luteoma, exogén 2. defect in action (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD 3. LH receptor mutation (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome
 1. Ovotesticular DSD. 2. Partial Gonadal Dysgenesis. 2. testis SRY+, SOX9 duplication. 3. Testicular regression. 3. gonadal dysgenesis. 4. Ovotesticular DSD. 47, XXY Klinefelter syndrome. Defect in androgen biosynthesis or in androgen action. Androgen overproduction. 1. defect in biosynthesis (17hydroxilase defect, 17 HSD, 5reductase defect, Star mutation) 1. fetal origin: 21-OH def./11OH def. 2. fetoplacental origin aromatase defect. 3. maternal origin: luteoma, exogén. 2. defect in action. (CAIS, PAIS) 45X/46,XY Mixed gonadal dysgenesis. (ovoteticular DSD) 46,XX/46,XY Chimeric ovotesticular DSD. 3. LH receptor mutation. (Leydig-hypoplasia, aplasia) 4. Persistent Müllerian duct syndrome.")
41
Treatment

42
TS

44
KS

45
17 -OH

46
CGD

47
CAIS

48
21 OH

49
PGD

50
PAIS

52
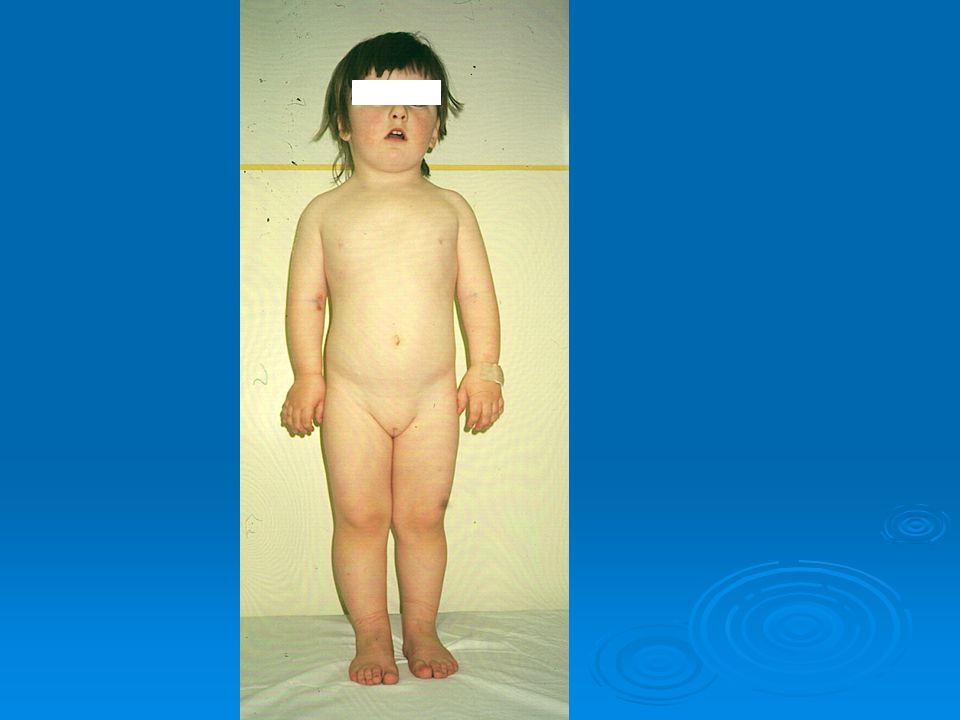
Rickets

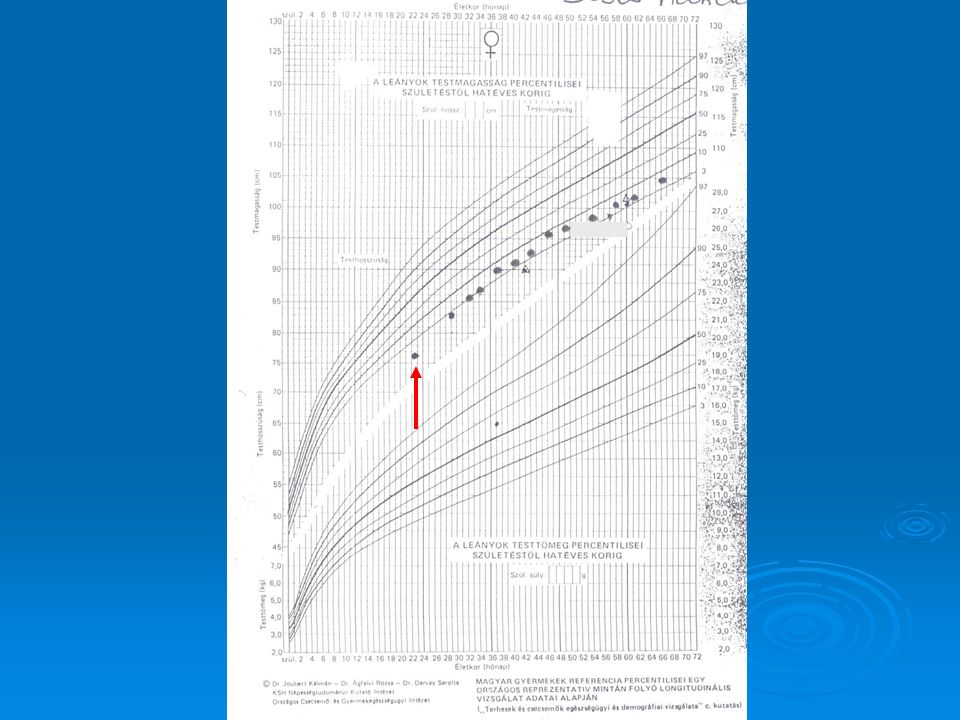
53
Definition Decrease in the enchondral calcification of the growth plate. Growth plate deformities Decreased growth rate Skeletal deformities In adults: osteomalacia

54
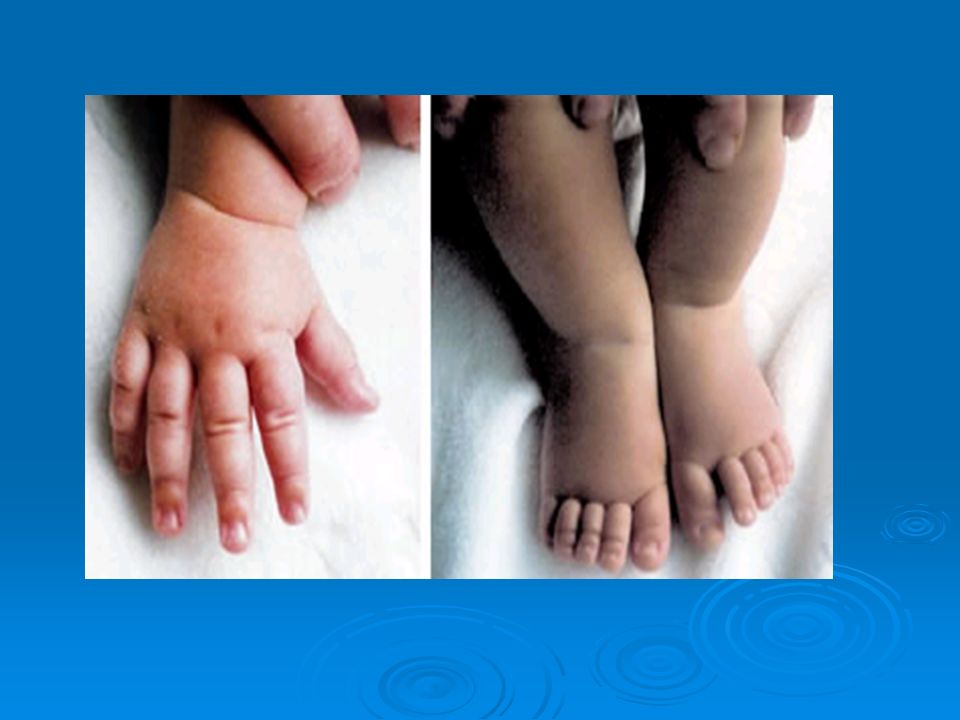
Clinical signs Decreased growth rate Big fontanelle, craniotabes
Swelling around growth plate: Rachitic bracelet

55
Bowleg (genu varum) windswept
(genu varum + genu valgum)
")
56
Rachitic rosary Harrison’s sulcus

57
Radiographic findings
Loss of demarcation Growth plate widens Irregular outlines Metaphysis cuped, flared

58
Classification Calciopenic Vitamin D-deficient
Vitamin D 1- hydrohylase deficiency Hereditary 1,25 (OH)2 D resistant Phospopenic X-linked hypophosphatemic AD hypophosphatemic Hereditary hypophosphatemic rickets with hypercalciuria Renal tubular disorder – Fanconi sy.
2 D resistant. Phospopenic. X-linked hypophosphatemic. AD hypophosphatemic. Hereditary hypophosphatemic rickets with hypercalciuria. Renal tubular disorder – Fanconi sy.")
59
Vitamin D- deficient rickets
Inadequate vitamin D intake or formation in the skin (UV B dependent) 3-18 moths of age Breatsfeeding high risk IU/ day
 3-18 moths of age. Breatsfeeding high risk IU/ day.")
60
Stages of vitamin D-deficient rickets
25(OH)D Calcium Low N PTH Starts to rise Phosphorus N low ALP
D. Calcium. Low N. PTH. Starts to rise. Phosphorus. N. low. ALP.")
61
Treatment Prevention 400-600 IU/ day
Vitamin D IU/ day – 6-8 weeks ( IU inramuscular) Ca supplementation: mg/ day Prevention IU/ day
 Ca supplementation: mg/ day. Prevention IU/ day.")
62
Vitamin D 1- hyroxylase deficiency Vitamin D dependent rickets type I
renal 1 - hydroxylase is inactive During the first 2 years of life Treatment 0,25-2,0 ug/day calcitriol + calcium

63
Hereditary 1,25 (OH)2- resistant rickets Vitamin D dependent rickets typeII
vitamin D receptor mutation- ressitance to vitamin D Treatment very high dose of calcitriol

64
X-linked hypophosphatemic rickets
PHEx (phosphate-regulating gene with homologie to endopeptidases) X kr. Renal phosphate wasting hypophospataemia inapproprate 1,25 (OH)2 D Treatment Elemental phosphorus + calcitriol
 X kr. Renal phosphate wasting. hypophospataemia. inapproprate 1,25 (OH)2 D. Treatment. Elemental phosphorus + calcitriol.")
65
AD hypophosphatemic rickets
Rare disorder FGF23 (fibroblast growth factor family) Presentation: childhood, adulthood
 Presentation: childhood, adulthood.")
66
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH)
Similar to XLH BUT: appropriate 1,25 (OH)2 Vit. D hypercalciuria – kidney stones Treatment Phosphorus supplementation
2 Vit. D. hypercalciuria – kidney stones. Treatment. Phosphorus supplementation.")
67
Renal tubular disosrders- Fanconi syndrome
Defects in the reabsorption of ion (Mg, P, Ca, Na, K, HCO3-), glucose, amino acids Causes: cystinosis, tyrosinaemia, galactosemia, Lowe syndrome Treatment phosphorus, calcitriol
, glucose, amino acids. Causes: cystinosis, tyrosinaemia, galactosemia, Lowe syndrome. Treatment. phosphorus, calcitriol.")
68
Biochemical finding in different forms of rickets
Serum Urine Ca P PTH ALP 25(OH)D 1,25(OH) TRP AAS Vit.D def N N/L H/N H L L/N/H - 1 def Vit.D res. HH Hypophos. ~+ hypercalciuria Fanconi +
D. 1,25(OH) TRP. AAS. Vit.D def. N. N/L. H/N. H. L. L/N/H. - 1 def. Vit.D res. HH. Hypophos. ~+ hypercalciuria. Fanconi. +")
Similar presentations




 become male or female?>")






>")











