Download presentation
Presentation is loading. Please wait.

1
Parkinson’s Disease Pathology 430/826 The Molecular Basis of Disease Neurological Genetics 9 th March 2015 Dr. John Rossiter rossiter@cliff.path.queensu.ca

7
Astroglial cells (Astrocytes)
")
8
Oligodendrocyte

10
Neurodegenerative Diseases A heterogeneous group of slowly progressive neurological diseases. Individual disorders traditionally classified on the basis of their clinical and pathological features. Characterised by selective loss of certain anatomically and/or physiologically related systems of neurons.

11
Some examples: Cerebral Cortex - Alzheimer’s disease - Pick’s disease -Basal Ganglia - Huntington’s disease - Parkinsons disease Motor System - ALS Spinocerebellar Syst. - Friedreich’s ataxia Peripheral nerves - Charcot-Marie-Tooth Note: the above list indicates regions of preferential, but not exclusive, neuronal loss. For example, in AD there is also degeneration and loss of neurons in subcortical structures and brainstem, and in HD there is also neuronal loss in the cerebral cortex.

12
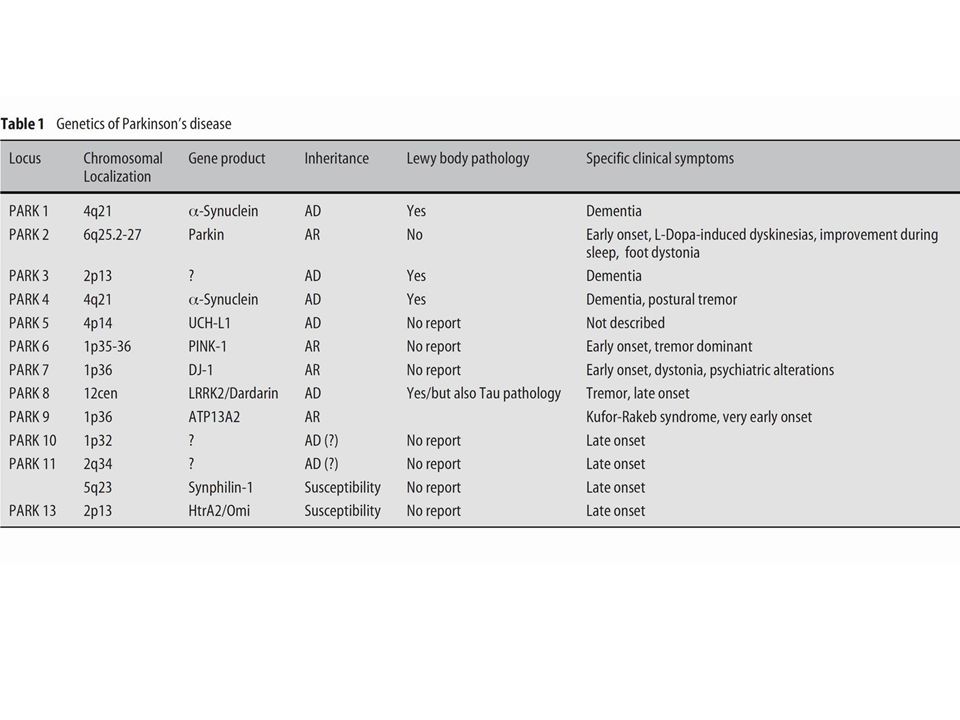
Disorders such as Alzheimer’s disease, Parkinson’s disease (and ALS) likely result from the interaction of a variety of age-related, genetic and environmental factors. These factors ‘converge’ to result in a similar clinico- pathological phenotype. Identification of gene mutations in rare familial cases gives important insights into the pathogenesis of the much commoner sporadic forms of these diseases.

13
Some important recurring themes in the pathogenesis of neurodegenerative diseases Increasing age is a major risk factor, likely in part related to defects in mitochondrial energy metabolism. Abnormal protein folding and aggregation appears to play a key pathogenetic role in many neurodegenerative diseases. Synaptic dysfunction is likely an important early event. Neuronal cell death does eventually occur, but neurons have likely been stressed and adaptive/maladaptive for months or years before this. Excitatory amino acid toxicity (excitotoxicity) is probably an important pathogenetic factor, especially in HD and ALS.
 is probably an important pathogenetic factor, especially in HD and ALS..")
14
J.J. Palop et al, Nature 19 Oct 2006, 768-773

15
A strong case can be made for understanding many Neurodegenerative diseases as: ‘Protein Aggregation Disorders’ Abnormal protein folding and resulting aggregation causes: Loss of normal protein function Gain of Toxic function

16
Dr. James Parkinson

19
Dopaminergic projection systems Blumenfeld Fig. 14.10

20
W Matsuda et al. J Neurosci. 14 Jan 2009

21
JD Surmeier et al., Neuroscience 198 (2011)221-231
")
24
Loss of pigmented dopaminergic neurons in the substantia nigra and a Lewy body inclusion in the cytoplasm of a surviving neuron

25
* * Two Lewy bodies * in the cytoplasm of a neuron in the pars compacta of the substantia nigra Nucleus

26
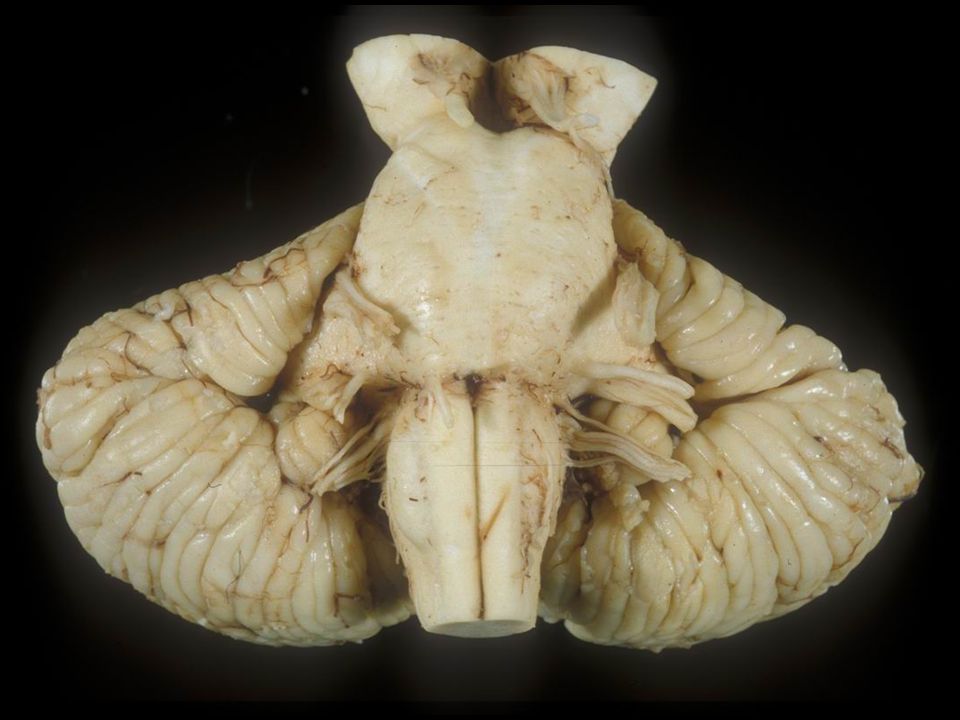
Noradrenergic projection systems Blumenfeld Fig. 14.11 Normal rostral ponsAdvanced PD The sections of the rostral pons above shows the normal locus bilaterally containing pigmented noradrenergic neurons and marked depigmentation of these nuclei in a case of advanced Parkinson’s disease (right panel).
..")
27
Mutations in the alpha-synuclein gene cause rare autosomal dominant cases of familial AD Science, 27 June 1997

29
Intense immunoreactivity of Lewy bodies for alpha-synuclein protein. Nature, Aug. 1997 In the common (sporadic) form of PD, Lewy bodies are formed predominantly from aggregates of alpha-synuclein (normal gene but abnormal protein metabolism)
 form of PD, Lewy bodies are formed predominantly from aggregates of alpha-synuclein (normal gene but abnormal protein metabolism).")
30
α-synuclein-immunoreactive Lewy bodies (blue arrows) and Lewy neurites (yellow arrows for representative examples ) in the midbrain tegmentum.
 and Lewy neurites (yellow arrows for representative examples ) in the midbrain tegmentum.")
34
α-synuclein Both mutations in alpha synuclein and overproduction of the normal protein can directly cause PD Role of alpha synuclein in the much commoner sporadic forms of PD ? A polymorphic microsatellite repeat in the alpha synuclein promoter associated with sporadic PD in some studies, but not in others

35
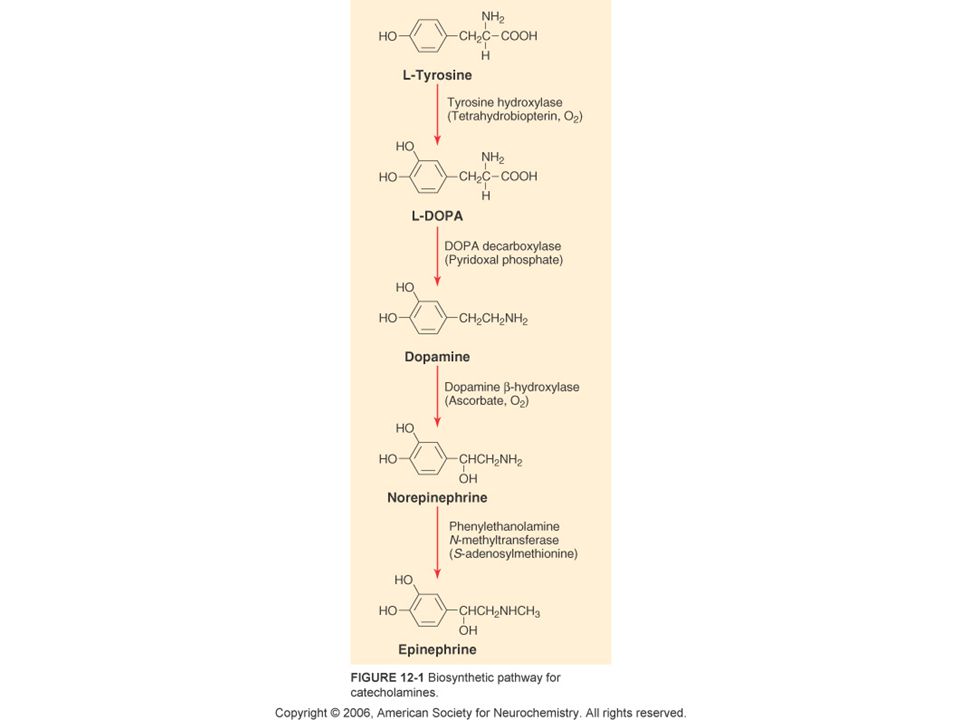
α-Synuclein α-synuclein is a small protein (140 amino acids). It is highly expressed in neurons, especially at presynaptic nerve endings. It has several normal functions, including the regulation of vesicle dynamics at presynaptic nerve endings and the fusion and fission of mitochondria. It also appears to have a regulatory role in autophagy. In Parkinson’s disease and other ‘Synucleinopathies’ individual α- synuclein molecules change their configuration and this promotes the formation of oligomers and subsequently insoluble polymers that form Lewy bodies and Lewy neurites. PD pathogenesis probably results from a combination of loss of normal function and gain of toxic function of α-synuclein, with monomers and oligomers likely being the most toxic form of altered α-synuclein.

39
Science, 21May 2004

40
B. Levine, Nature, 2007; 446:746

45
In ‘Diffuse Lewy body disease’ / ‘Dementia with Lewy bodies’, Lewy bodies and neurites are found more extensively distributed in the CNS, especially in the cerebral cortex. Third report of the DLB consortium. Neurology 65 (2005) 1863-71
")
46
H&E stainUbiquitin immunostain α-synuclein-immunostain Cortical Lewy bodies in a case of Diffuse Lewy body disease * *

47
Cortical Lewy bodies are only the ‘tip of the iceberg’. A sensitive immuno- detection technique shows diffuse α-synuclein aggregation in the cerebral cortex of a DLB case versus normal brain. Kramer et al., J. Neuroscience, Feb 2007

53
Dr. James Parkinson

Similar presentations






















 A 42 deposits subtle.>")







 By Abdelkader Ashour, Ph.D. Phone: 4677212>")










