Download presentation
Presentation is loading. Please wait.

1
Computational Biology, Part 10 Protein Structure Prediction and Display Robert F. Murphy Copyright 1996, 1999, 2001. All rights reserved.

2
Goal Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each residue Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each residue
 and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each residue Take primary structure (sequence) and, using rules derived from known structures, predict the secondary structure that is most likely to be adopted by each residue")
3
Structural Propensities Due to the size, shape and charge of its side chain, each amino acid may “fit” better in one type of secondary structure than another Due to the size, shape and charge of its side chain, each amino acid may “fit” better in one type of secondary structure than another Classic example: The rigidity and side chain angle of proline cannot be accomodated in an -helical structure Classic example: The rigidity and side chain angle of proline cannot be accomodated in an -helical structure

4
Structural Propensities Two ways to view the significance of this preference (or propensity) Two ways to view the significance of this preference (or propensity) It may control or affect the folding of the protein in its immediate vicinity (amino acid determines structure) It may constitute selective pressure to use particular amino acids in regions that must have a particular structure (structure determines amino acid)
 Two ways to view the significance of this preference (or propensity) It may control or affect the folding of the protein in its immediate vicinity (amino acid determines structure) It may constitute selective pressure to use particular amino acids in regions that must have a particular structure (structure determines amino acid)")
5
Secondary structure prediction In either case, amino acid propensities should be useful for predicting secondary structure In either case, amino acid propensities should be useful for predicting secondary structure Two classical methods that use previously determined propensities: Two classical methods that use previously determined propensities: Chou-Fasman Garnier-Osguthorpe-Robson

6
Chou-Fasman method Uses table of conformational parameters (propensities) determined primarily from measurements of secondary structure by CD spectroscopy Uses table of conformational parameters (propensities) determined primarily from measurements of secondary structure by CD spectroscopy Table consists of one “likelihood” for each structure for each amino acid Table consists of one “likelihood” for each structure for each amino acid
 determined primarily from measurements of secondary structure by CD spectroscopy Uses table of conformational parameters (propensities) determined primarily from measurements of secondary structure by CD spectroscopy Table consists of one likelihood for each structure for each amino acid Table consists of one likelihood for each structure for each amino acid")
7
Chou-Fasman propensities (partial table)
")
8
Chou-Fasman method A prediction is made for each type of structure for each amino acid A prediction is made for each type of structure for each amino acid Can result in ambiguity if a region has high propensities for both helix and sheet (higher value usually chosen, with exceptions)
")
9
Chou-Fasman method Calculation rules are somewhat ad hoc Calculation rules are somewhat ad hoc Example: Method for helix Example: Method for helix Search for nucleating region where 4 out of 6 a.a. have P > 1.03 Extend until 4 consecutive a.a. have an average P < 1.00 If region is at least 6 a.a. long, has an average P > 1.03, and average P > average P consider region to be helix

10
Garnier-Osguthorpe-Robson Uses table of propensities calculated primarily from structures determined by X- ray crystallography Uses table of propensities calculated primarily from structures determined by X- ray crystallography Table consists of one “likelihood” for each structure for each amino acid for each position in a 17 amino acid window Table consists of one “likelihood” for each structure for each amino acid for each position in a 17 amino acid window

11
Garnier-Osguthorpe-Robson Analogous to searching for “features” with a 17 amino acid wide frequency matrix Analogous to searching for “features” with a 17 amino acid wide frequency matrix One matrix for each “feature” One matrix for each “feature” -helix -sheet turn coil Highest scoring “feature” is found at each location Highest scoring “feature” is found at each location

12
Accuracy of predictions Both methods are only about 55-65% accurate Both methods are only about 55-65% accurate A major reason is that while they consider the local context of each sequence element, they do not consider the global context of the sequence - the type of protein A major reason is that while they consider the local context of each sequence element, they do not consider the global context of the sequence - the type of protein The same amino acids may adopt a different configuration in a cytoplasmic protein than in a membrane protein

13
“Adaptive” methods Neural network methods - train network using sets of known proteins then use to predict for query sequence Neural network methods - train network using sets of known proteins then use to predict for query sequence nnpredict Homology-based methods - predict structure using rules derived only from proteins homologous to query sequence Homology-based methods - predict structure using rules derived only from proteins homologous to query sequence SOPM PHD

14
Neural Network methods A neural network with multiple layers is presented with known sequences and structures - network is trained until it can predict those structures given those sequences A neural network with multiple layers is presented with known sequences and structures - network is trained until it can predict those structures given those sequences Allows network to adapt as needed (it can consider neighboring residues like GOR) Allows network to adapt as needed (it can consider neighboring residues like GOR)
 Allows network to adapt as needed (it can consider neighboring residues like GOR)")
15
Neural Network methods Different networks can be created for different types of proteins Different networks can be created for different types of proteins

16
Homology-based modeling Principle: From the sequences of proteins whose structures are known, choose a subset that is similar to the query sequence Principle: From the sequences of proteins whose structures are known, choose a subset that is similar to the query sequence Develop rules (e.g., train a network) for just this subset Develop rules (e.g., train a network) for just this subset Use these rules to make prediction for the query sequence Use these rules to make prediction for the query sequence
 for just this subset Develop rules (e.g., train a network) for just this subset Use these rules to make prediction for the query sequence Use these rules to make prediction for the query sequence")
17
Retrieving 3D structures Protein Data Bank (PDB) Protein Data Bank (PDB) using web browser home page = http://www.pdb.bnl.gov/ using anonymous FTP Entrez Entrez using web browser BLAST BLAST using web browser
 Protein Data Bank (PDB) using web browser home page = using anonymous FTP Entrez Entrez using web browser BLAST BLAST using web browser")
18
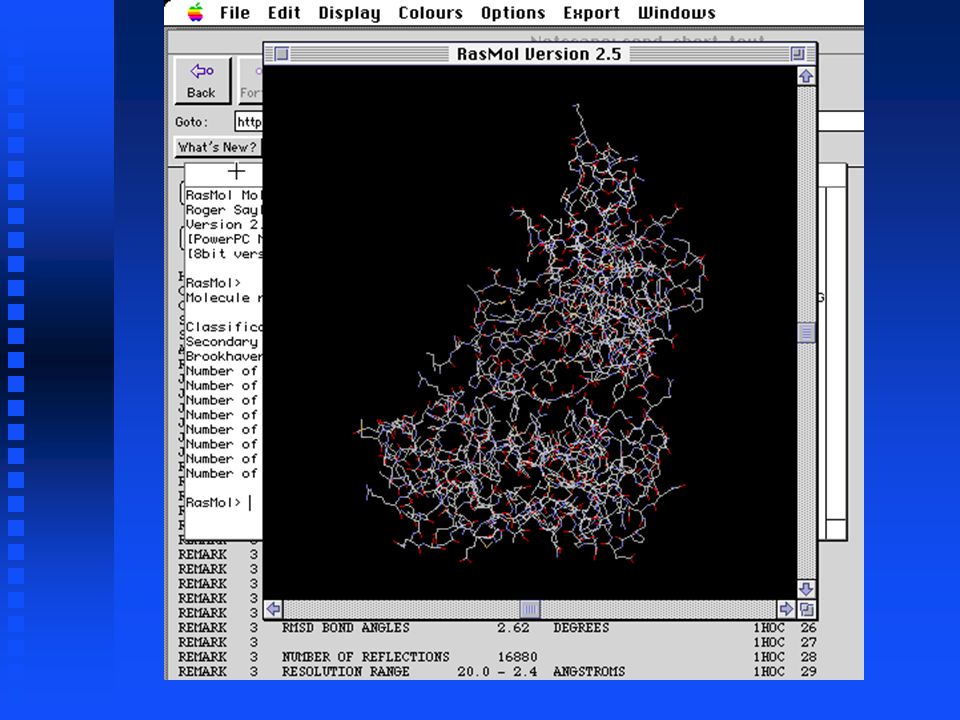
Displaying Structures with RasMol The GIF image of Ribonuclease A is static - we cannot rotate the molecule or recolor portions of it to aid visualization The GIF image of Ribonuclease A is static - we cannot rotate the molecule or recolor portions of it to aid visualization For this we can use RasMol, a public domain program available for wide range of computers, including MacOS, Windows and Unix For this we can use RasMol, a public domain program available for wide range of computers, including MacOS, Windows and Unix

19
Displaying Structures with RasMol Drs. David Hackney and Will McClure have developed an online tutorial for RasMol - a link may be found on the 03-310, 03-311 and 03-510 web pages Drs. David Hackney and Will McClure have developed an online tutorial for RasMol - a link may be found on the 03-310, 03-311 and 03-510 web pages

20
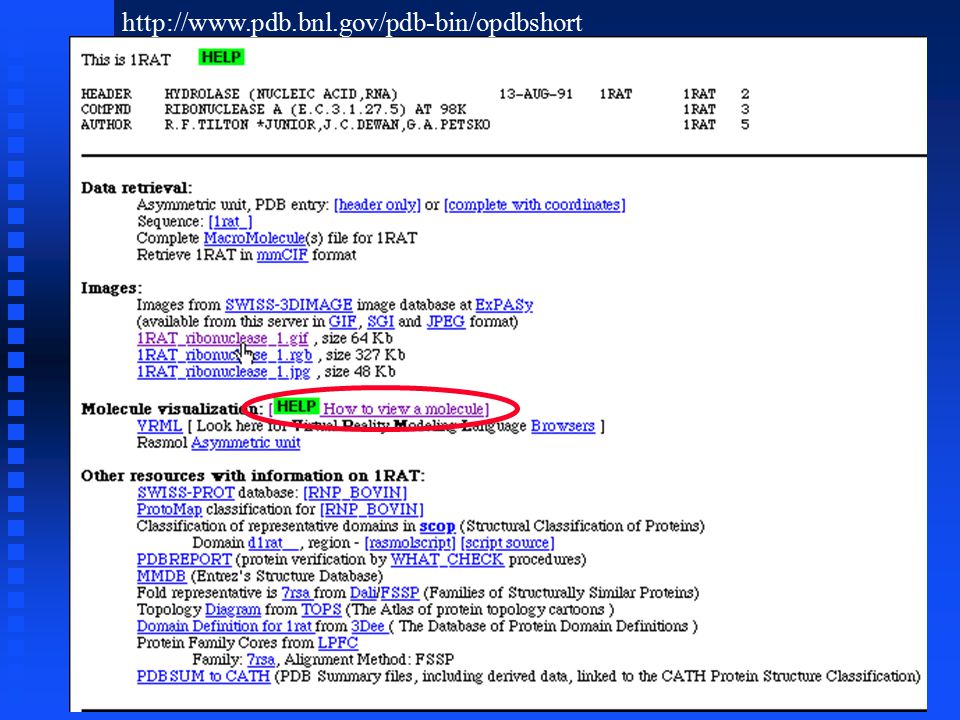
PDB files In order to optimally display, rotate and color the 3D structure, we need to download a copy of the coordinates for each atom in the molecule to our local computer In order to optimally display, rotate and color the 3D structure, we need to download a copy of the coordinates for each atom in the molecule to our local computer The most common format for storage and exchange of atomic coordinates for biological molecules is PDB file format The most common format for storage and exchange of atomic coordinates for biological molecules is PDB file format

21
PDB files PDB file format is a text (ASCII) format, with an extensive header that can be read and interpreted either by programs or by people PDB file format is a text (ASCII) format, with an extensive header that can be read and interpreted either by programs or by people We can request either the header only or the entire file; the next screen requests the header only We can request either the header only or the entire file; the next screen requests the header only
 format, with an extensive header that can be read and interpreted either by programs or by people PDB file format is a text (ASCII) format, with an extensive header that can be read and interpreted either by programs or by people We can request either the header only or the entire file; the next screen requests the header only We can request either the header only or the entire file; the next screen requests the header only")
22
http://www.pdb.bnl.gov/pdb-bin/opdbshort

23
http://www.pdb.bnl.gov/pdb-bin/send-pdb?filename=1rat&short=1

24
http://www.pdb.bnl.gov/pdb-bin/opdbshort
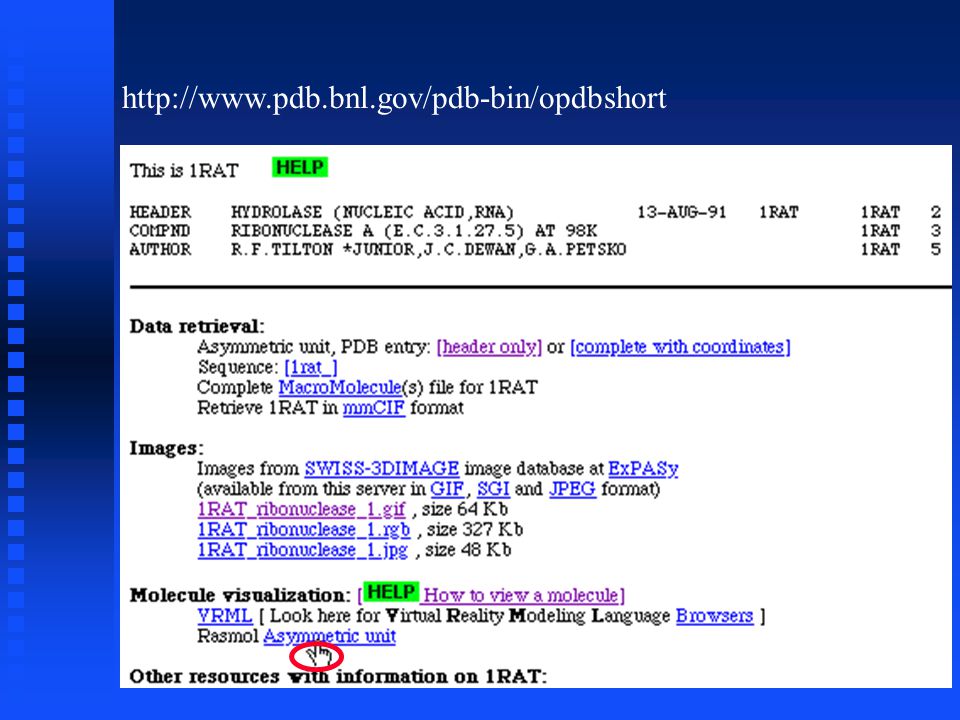
25
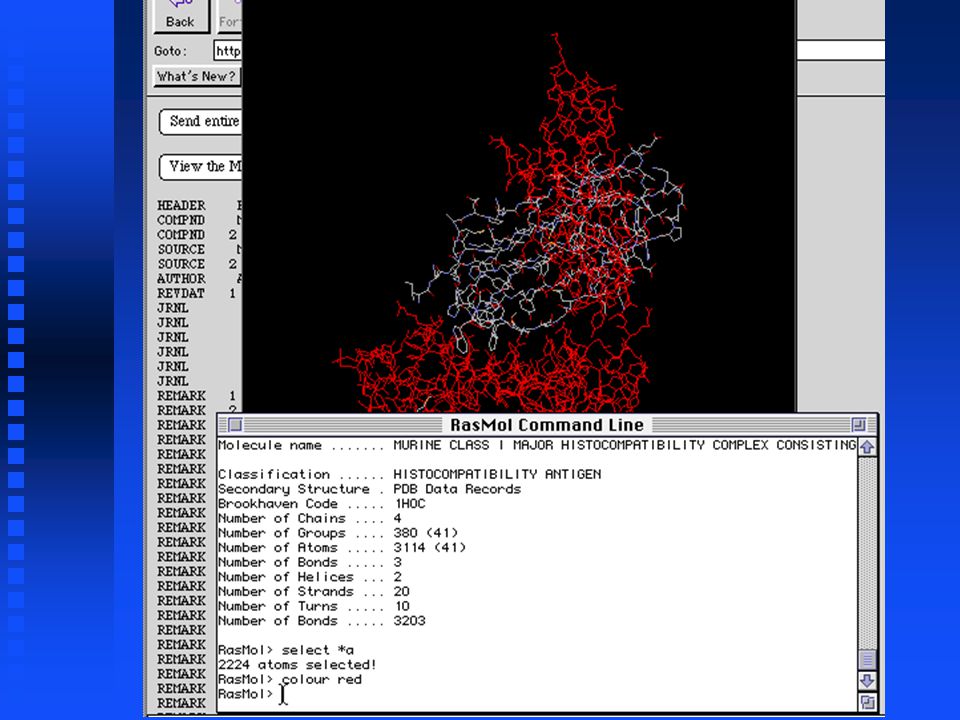
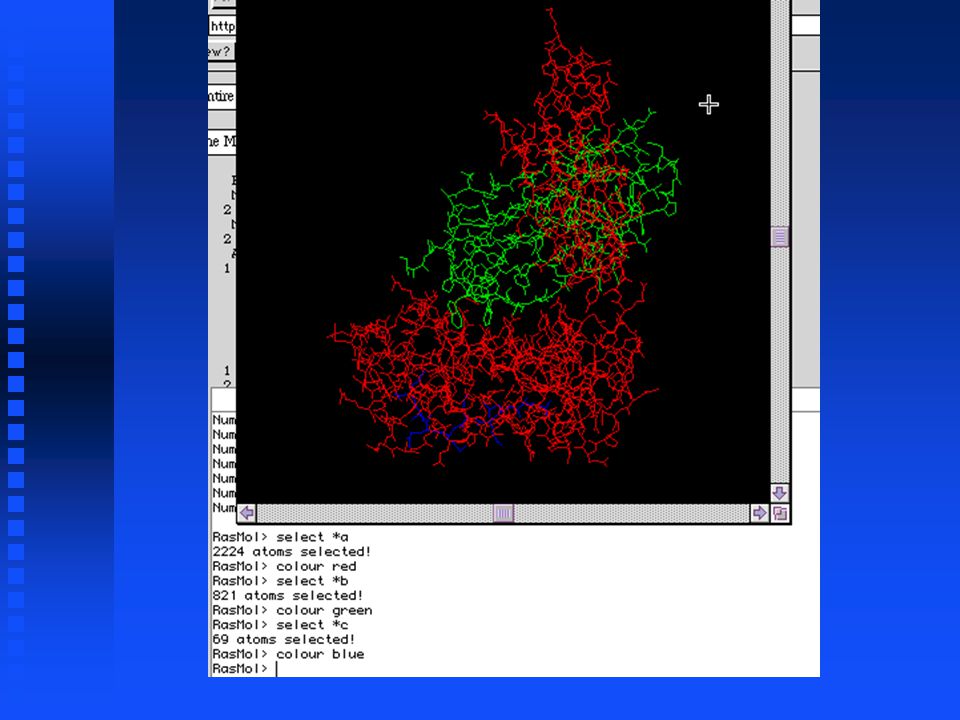
RasMol has a graphics window and a command window

26
PDB Retrieval & Display Can download PDB files from Entrez Can download PDB files from Entrez Second example: Display structures of MHC proteins containing 2 -microglobulin Second example: Display structures of MHC proteins containing 2 -microglobulin

28
Useful RasMol commands show sequence lists all amino acids in each chain show sequence lists all amino acids in each chain select *a selects all residues in chain A select *a selects all residues in chain A colour red displays the selected residues in red colour red displays the selected residues in red

31
Alternatives to RasMol NCBI (providers of Entrez service) have developed a public domain 3D viewer for molecules, Cn3D (“See in 3D”) NCBI (providers of Entrez service) have developed a public domain 3D viewer for molecules, Cn3D (“See in 3D”) Integrated into Network Entrez Client Integrated into Network Entrez Client Available as a stand-alone helper application Available as a stand-alone helper application
 have developed a public domain 3D viewer for molecules, Cn3D ( See in 3D ) NCBI (providers of Entrez service) have developed a public domain 3D viewer for molecules, Cn3D ( See in 3D ) Integrated into Network Entrez Client Integrated into Network Entrez Client Available as a stand-alone helper application Available as a stand-alone helper application")
32
Alternatives to RasMol It is often useful for an investigator or teacher to be able to save a series of views of one or more molecules so that they can be replayed again (creating a script for a “movie” with preprogrammed changes in rotation, color, etc.) It is often useful for an investigator or teacher to be able to save a series of views of one or more molecules so that they can be replayed again (creating a script for a “movie” with preprogrammed changes in rotation, color, etc.) Two programs that do this are CHIME and MAGE Two programs that do this are CHIME and MAGE
 It is often useful for an investigator or teacher to be able to save a series of views of one or more molecules so that they can be replayed again (creating a script for a movie with preprogrammed changes in rotation, color, etc.) Two programs that do this are CHIME and MAGE Two programs that do this are CHIME and MAGE")
33
Alternatives to RasMol CHIME (derived from RasMol source) is available as a Browser Plugin CHIME (derived from RasMol source) is available as a Browser Plugin MAGE is available as a stand-alone helper application MAGE is available as a stand-alone helper application Information on both is available through links on a HELP page at the PDB Information on both is available through links on a HELP page at the PDB
 is available as a Browser Plugin CHIME (derived from RasMol source) is available as a Browser Plugin MAGE is available as a stand-alone helper application MAGE is available as a stand-alone helper application Information on both is available through links on a HELP page at the PDB Information on both is available through links on a HELP page at the PDB")
34
http://www.pdb.bnl.gov/pdb-bin/opdbshort
35
Structural homology It is useful for new proteins whose 3D structure is not known to be able to find proteins whose 3D structure is known that are expected to have a similar structure to the unknown It is useful for new proteins whose 3D structure is not known to be able to find proteins whose 3D structure is known that are expected to have a similar structure to the unknown It is also useful for proteins whose 3D structure is known to be able to find other proteins with similar structures It is also useful for proteins whose 3D structure is known to be able to find other proteins with similar structures

36
Finding proteins with known structures based on sequence homology If you want to find known 3D structures of proteins that are similar in primary amino acid sequence to a particular sequence, can use BLAST web page and choose the PDB database If you want to find known 3D structures of proteins that are similar in primary amino acid sequence to a particular sequence, can use BLAST web page and choose the PDB database This is not the PDB database of structures, rather a database of amino acid sequences for those proteins in the structure database This is not the PDB database of structures, rather a database of amino acid sequences for those proteins in the structure database Links are available to retrieve PDB files Links are available to retrieve PDB files

37
Finding proteins with similar structures to a known protein For literature and sequence databases, Entrez allows neighbors to be found for a selected entry based on “homology” in terms (MEDline database) or sequence (protein and nucleic acid sequence databases) For literature and sequence databases, Entrez allows neighbors to be found for a selected entry based on “homology” in terms (MEDline database) or sequence (protein and nucleic acid sequence databases) An experimental feature allows neighbors to be chosen for entries in the structure database An experimental feature allows neighbors to be chosen for entries in the structure database
 or sequence (protein and nucleic acid sequence databases) For literature and sequence databases, Entrez allows neighbors to be found for a selected entry based on homology in terms (MEDline database) or sequence (protein and nucleic acid sequence databases) An experimental feature allows neighbors to be chosen for entries in the structure database An experimental feature allows neighbors to be chosen for entries in the structure database")
38
Finding proteins with similar structures to a known protein Proteins with similar structures are termed “VAST Neighbors” by Entrez (VAST refers to the method used to evaluate similarity of structure) Proteins with similar structures are termed “VAST Neighbors” by Entrez (VAST refers to the method used to evaluate similarity of structure) VAST or structure neighbors may or may not have sequence homology to each other VAST or structure neighbors may or may not have sequence homology to each other
 Proteins with similar structures are termed VAST Neighbors by Entrez (VAST refers to the method used to evaluate similarity of structure) VAST or structure neighbors may or may not have sequence homology to each other VAST or structure neighbors may or may not have sequence homology to each other")
Similar presentations






![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/15/4859888/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")



![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")

>")
 Protein Structure Prediction Protein Secondary Structure.>")


![Protein structure determination & prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray.](/16/5177546/big_thumb.jpg "Protein structure determination & prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray.>")


