Download presentation
Presentation is loading. Please wait.

1
Database searching

2
Purposes of similarity search Function prediction by homology (in silico annotation) Function prediction by homology (in silico annotation) Search for identified gene in other organisms Search for identified gene in other organisms Identifying regulatory elements Identifying regulatory elements Assisting in sequence assembly Assisting in sequence assemblyProblems Similar sequences can have different functions Similar sequences can have different functions Non-homologous sequences can have identical function Non-homologous sequences can have identical function Feature space <> Sequence space Feature space <> Sequence space
 Function prediction by homology (in silico annotation) Search for identified gene in other organisms Search for identified gene in other organisms Identifying regulatory elements Identifying regulatory elements Assisting in sequence assembly Assisting in sequence assemblyProblems Similar sequences can have different functions Similar sequences can have different functions Non-homologous sequences can have identical function Non-homologous sequences can have identical function Feature space <> Sequence space Feature space <> Sequence space")
3
Some databases nr (GenBank nucleotide and protein) nr (GenBank nucleotide and protein) nr Month: monthly update Month: monthly update swissprot (protein) swissprot (protein) swissprot pdb (proteins with 3D structures) pdb (proteins with 3D structures) pdb Various genome databases (human, mouse etc) Various genome databases (human, mouse etc)
 nr (GenBank nucleotide and protein) nr Month: monthly update Month: monthly update swissprot (protein) swissprot (protein) swissprot pdb (proteins with 3D structures) pdb (proteins with 3D structures) pdb Various genome databases (human, mouse etc) Various genome databases (human, mouse etc)")
4
Main tools FASTA FASTA BLAST=Basic Local Alignment Search Tool BLAST=Basic Local Alignment Search ToolProcedure 1. Choose scoring matrix 2. Find best local alignments using scoring matrix 3. Determine statistical significance of result List in decreasing order of significance List in decreasing order of significance

5
Blosum substitution matrix log odds scores 2log(proportion observed/proportion expected)
")
6
FASTA Step 1 : Find hot-spots Step 1 : Find hot-spots (i.e. pairs of words of length k) that exactly match. (hashing) Step 2: Locate best “diagonal runs”(sequences of consecutive hot spots on a diagonal) Step 2: Locate best “diagonal runs”(sequences of consecutive hot spots on a diagonal) Step 3 : Combine sub-alignments Step 3 : Combine sub-alignments form diagonal runs into a longer alignment
 that exactly match. (hashing) Step 2: Locate best diagonal runs (sequences of consecutive hot spots on a diagonal) Step 2: Locate best diagonal runs (sequences of consecutive hot spots on a diagonal) Step 3 : Combine sub-alignments Step 3 : Combine sub-alignments form diagonal runs into a longer alignment.")
7
Exercise (hashing Tables of FASTA) sequence 1: ACNGTSCHQE sequence 2: GCHCLSAGQD Prepare Table of offset values = matching diagonals
 sequence 1: ACNGTSCHQE sequence 2: GCHCLSAGQD Prepare Table of offset values = matching diagonals")
8
Solution sequence 1: ACNGTSCHQE sequence 2: GCHCLSAGQD sequence 1: ACNGTSCHQE C S Q <<offset = 0 sequence 2: GCHCLSAGQD sequence 1: ACNGTSCHQE--- G C <<offset = -3 sequence 2: ---GCHCLSAGQD sequence 1: ACNGTSCHQE----- CH <<offset = -5 sequence 2: -----GCHCLSAGQD S T

9
The main steps of gapped BLAST 1. Specify word length (3 for proteins, 11 for nucleotides) 2. Filtering for complexity 3. Make list of words to search for 4. Exact search 5. Join matches, and extend ungapped alignment 6. Calculate E-values 7. Join high-scoring pairs 8. Perform Smith-Waterman on best matches

10
Filtering sequences Removing sequence regions of low complexity K Remove repeated elements (REPEATMASKER) Find K for sequence GGGG and for sequence ATCG
 Find K for sequence GGGG and for sequence ATCG")
11
The BLAST search algorithm

12
Determining significance of match The score of an ungapped alignment is The score of an ungapped alignment is S = sum s(x i,y i ). The scores of individual sites are independent. The scores of individual sites are independent. The distribution of the sum of independent random variables is a normal distribution (central limit theorem). The distribution of the sum of independent random variables is a normal distribution (central limit theorem).
. The distribution of the sum of independent random variables is a normal distribution (central limit theorem)..")
13
Determining significance of match However, we don't select scores randomly. We take the maximum extension of the initial word (HSP). However, we don't select scores randomly. We take the maximum extension of the initial word (HSP). The distribution of the maximum score of a large number N of i.i.d. random variables is called the extreme value distribution. The distribution of the maximum score of a large number N of i.i.d. random variables is called the extreme value distribution. P(Score greater than x)= Probability of observing a score S > x m’ and n’ are effective query and database lengths; K and l are substitution matrix parameters.
. However, we don t select scores randomly. We take the maximum extension of the initial word (HSP). The distribution of the maximum score of a large number N of i.i.d. random variables is called the extreme value distribution. The distribution of the maximum score of a large number N of i.i.d. random variables is called the extreme value distribution. P(Score greater than x)= Probability of observing a score S > x m’ and n’ are effective query and database lengths; K and l are substitution matrix parameters..")
14
Determining significance of match E-value = expected number of sequences scoring above S in the given database E-value = expected number of sequences scoring above S in the given database BIT-score: Sum of scores for local alignments BIT-score: Sum of scores for local alignments

15
Smith-Waterman local alignment

16
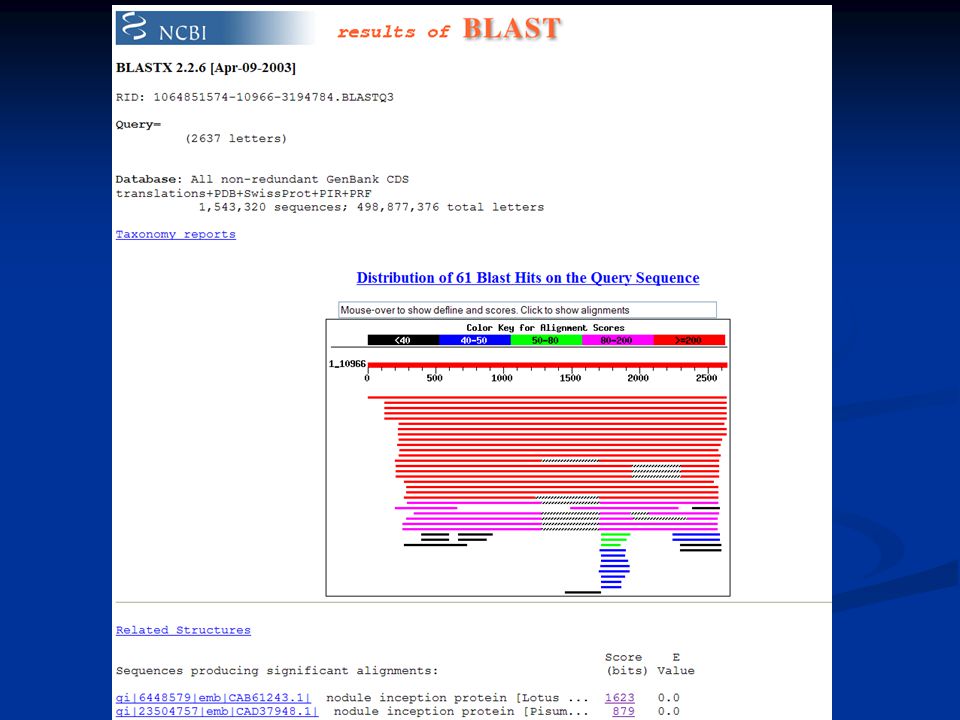
Example – nodulation Cloned sequence from Lotus japonicus Amino-acid level (BlastP) Amino-acid level (BlastP)BlastP LLANGNFVLRESGNKDQDGLVWQSFDFPTDTLLPQMKLGWDRKTGLNKI LRSWKSPSDPSSGYYSYKLEFQGLPEYFLNNRDSPTHRSGPWDGIRFSGIPEK Nucleotide level (BlastN) Nucleotide level (BlastN)BlastN cttctcgcta atggcaattt cgtgctaaga gagtctggca acaaagatca agatgggtta gtgtggcaga gtttcgattt tcccactgac actttactcc cgcagatgaa actgggatgg gatcgcaaaa cagggcttaa caaaatcctc agatcctgga aaagcccaag tgatccgtcaagtgggtatt actcgtataa actcgaattt caagggctcc ctgagtattt tttaaacaac agagactcgc caactcaccg gagcggtccg tgggatggta tccgatttag tggtattcca
 Amino-acid level (BlastP)BlastP LLANGNFVLRESGNKDQDGLVWQSFDFPTDTLLPQMKLGWDRKTGLNKI LRSWKSPSDPSSGYYSYKLEFQGLPEYFLNNRDSPTHRSGPWDGIRFSGIPEK Nucleotide level (BlastN) Nucleotide level (BlastN)BlastN cttctcgcta atggcaattt cgtgctaaga gagtctggca acaaagatca agatgggtta gtgtggcaga gtttcgattt tcccactgac actttactcc cgcagatgaa actgggatgg gatcgcaaaa cagggcttaa caaaatcctc agatcctgga aaagcccaag tgatccgtcaagtgggtatt actcgtataa actcgaattt caagggctcc ctgagtattt tttaaacaac agagactcgc caactcaccg gagcggtccg tgggatggta tccgatttag tggtattcca")
24
BLAST flavours Basic flavours Basic flavours BLASTP (proteins to protein database) BLASTP (proteins to protein database) BLASTN (nucleotides to nucleotide database) BLASTN (nucleotides to nucleotide database) BLASTX (translated nucleotides to protein database) BLASTX (translated nucleotides to protein database) TBLASTN (protein to translated database) TBLASTN (protein to translated database) TBLASTX (translated nucleotides to translated database) - SLOW TBLASTX (translated nucleotides to translated database) - SLOW
 BLASTP (proteins to protein database) BLASTN (nucleotides to nucleotide database) BLASTN (nucleotides to nucleotide database) BLASTX (translated nucleotides to protein database) BLASTX (translated nucleotides to protein database) TBLASTN (protein to translated database) TBLASTN (protein to translated database) TBLASTX (translated nucleotides to translated database) - SLOW TBLASTX (translated nucleotides to translated database) - SLOW")
25
Iterated searches Advanced family searches PSI-BLAST (Position Specific Iterated BLAST) PSI-BLAST (Position Specific Iterated BLAST)
 PSI-BLAST (Position Specific Iterated BLAST)")
26
PSI-blast Search with BLAST using the given query. Search with BLAST using the given query. while (there are new significant hits) while (there are new significant hits) combine all significant hits into a profile combine all significant hits into a profile search with BLAST using the profile search with BLAST using the profile end end
 while (there are new significant hits) combine all significant hits into a profile combine all significant hits into a profile search with BLAST using the profile search with BLAST using the profile end end.")
27
PSI-BLAST Greedy algorithm

28
Speeding up BLAST TeraBLAST on deCypher Hard-wiring the BLAST algorithm Hard-wiring the BLAST algorithm Up to 6 trillion BLASTN comparisons per second per DeCypher node. Up to 6 trillion BLASTN comparisons per second per DeCypher node. Up to 1 trillion amino acid comparisons per second per DeCypher node. Up to 1 trillion amino acid comparisons per second per DeCypher node. A single multi-node equipment rack replaces over 1,000 CPUs of BLAST throughput. A single multi-node equipment rack replaces over 1,000 CPUs of BLAST throughput.

29
TeraBLAST on deCypher Example: Human Genome Self-Similarity Search Example: Human Genome Self-Similarity Search (query x target)/2 = [3.4x10 9 ] 2 /2 = 5.8x10 18 base pair comparisons Search time for CPU server, software-BLASTN: Search time for CPU server, software-BLASTN: 5.8x10 18 /2x10 10 = 290 million seconds; or ~9.2 years. 5.8x10 18 /2x10 10 = 290 million seconds; or ~9.2 years. Search time for smallest DeCypher using Tera- BLASTN: 5.8x10 18 /2x10 12 = 2.9 million seconds; or ~33 days. Search time for smallest DeCypher using Tera- BLASTN: 5.8x10 18 /2x10 12 = 2.9 million seconds; or ~33 days.
![TeraBLAST on deCypher Example: Human Genome Self-Similarity Search Example: Human Genome Self-Similarity Search (query x target)/2 = [3.4x10 9 ] 2 /2 = 5.8x10 18 base pair comparisons Search time for CPU server, software-BLASTN: Search time for CPU server, software-BLASTN: 5.8x10 18 /2x10 10 = 290 million seconds; or ~9.2 years.](http://images.slideplayer.com/16/5027020/slides/slide_29.jpg "5.8x10 18 /2x10 10 = 290 million seconds; or ~9.2 years. Search time for smallest DeCypher using Tera- BLASTN: 5.8x10 18 /2x10 12 = 2.9 million seconds; or ~33 days. Search time for smallest DeCypher using Tera- BLASTN: 5.8x10 18 /2x10 12 = 2.9 million seconds; or ~33 days..")
30
Other genome projects Synteny of mouse chr 16 with the human genome (Science 2002) Synteny of mouse chr 16 with the human genome (Science 2002)
 Synteny of mouse chr 16 with the human genome (Science 2002)")
Similar presentations







 Species-specific databases Protein sequence GenBank (Entrez protein) UniProtKB (SwissProt) Protein structure.>")




 Function prediction by homology (in silico annotation)>")







 Function prediction by homology (in silico annotation)>")
 Function prediction by homology (in silico annotation)>")



