Download presentation
Presentation is loading. Please wait.

1
“Challenging” internal loop motifs Ali Mokdad, M.D., Ph.D.

4
Systematically finding internal loops

6
The state-of-the-art RNA automatic alignment methods are based on SCFG (covariance models) and do not systematically use all available 3D structural information for alignment. The advantage of using SCFG is their capability to describe nested interactions (RNA 2D structures). These methods as they are currently applied work best for helical W.C. segments, but do not produce accurate alignments in non helical segments or in areas where tertiary interactions occur. With the ever growing library of accurate RNA 3D structures, it is now possible to use the 3D information to build better alignments. Problem with current automatic alignment methods
. These methods as they are currently applied work best for helical W.C. segments, but do not produce accurate alignments in non helical segments or in areas where tertiary interactions occur. With the ever growing library of accurate RNA 3D structures, it is now possible to use the 3D information to build better alignments. Problem with current automatic alignment methods.")
7
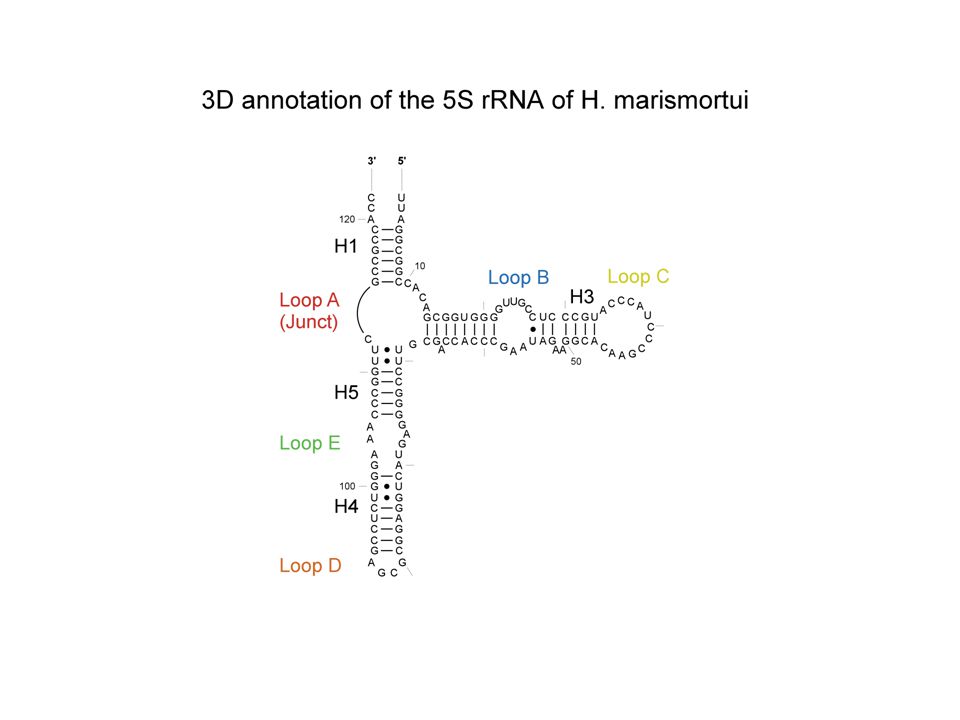
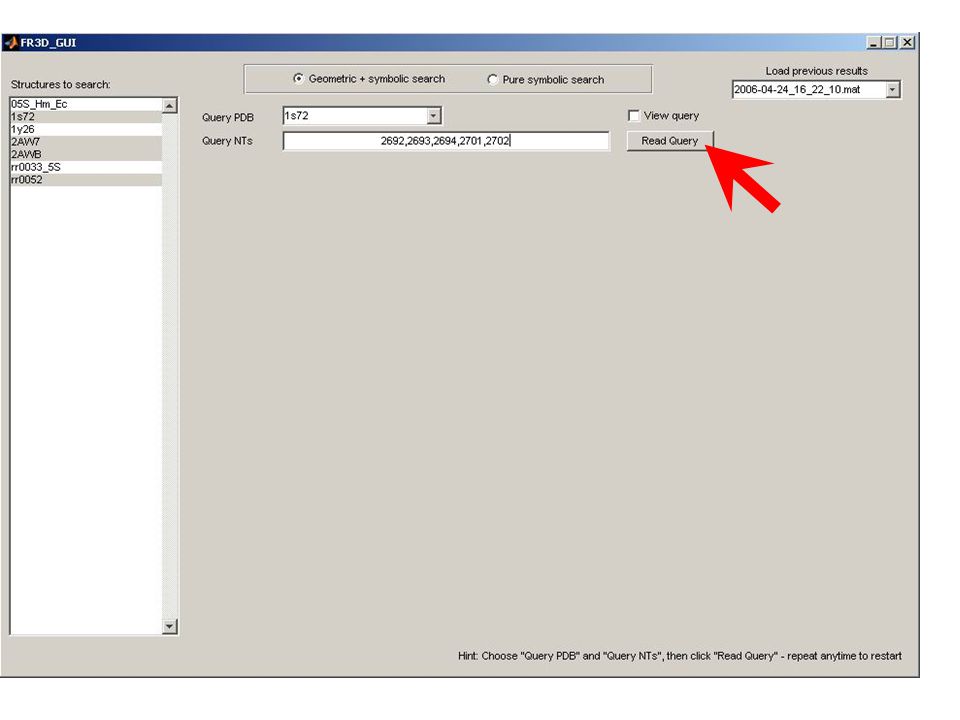
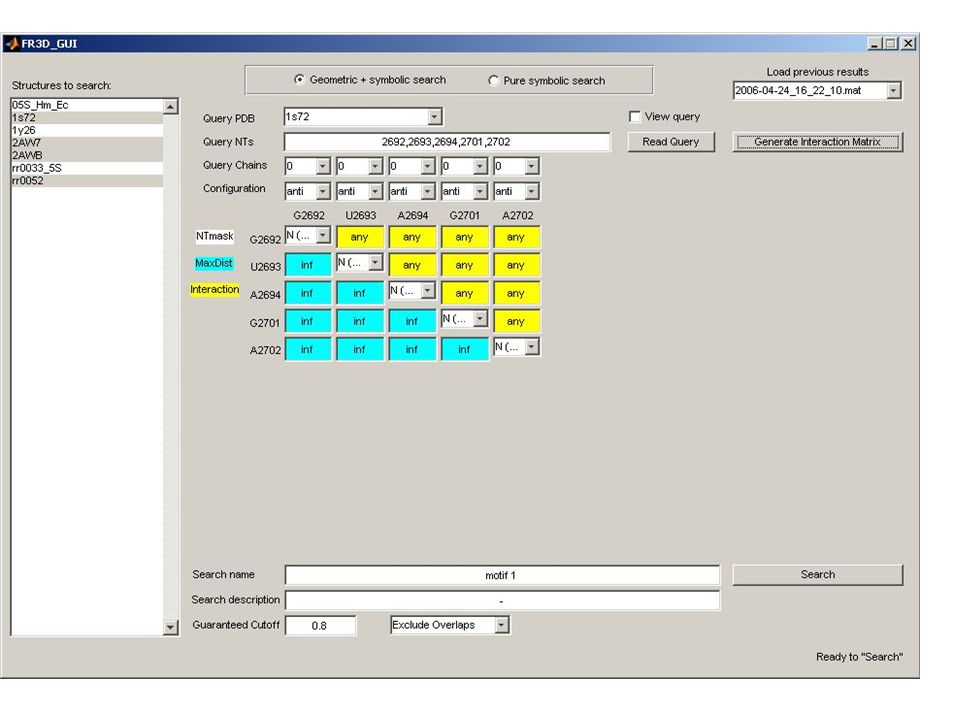
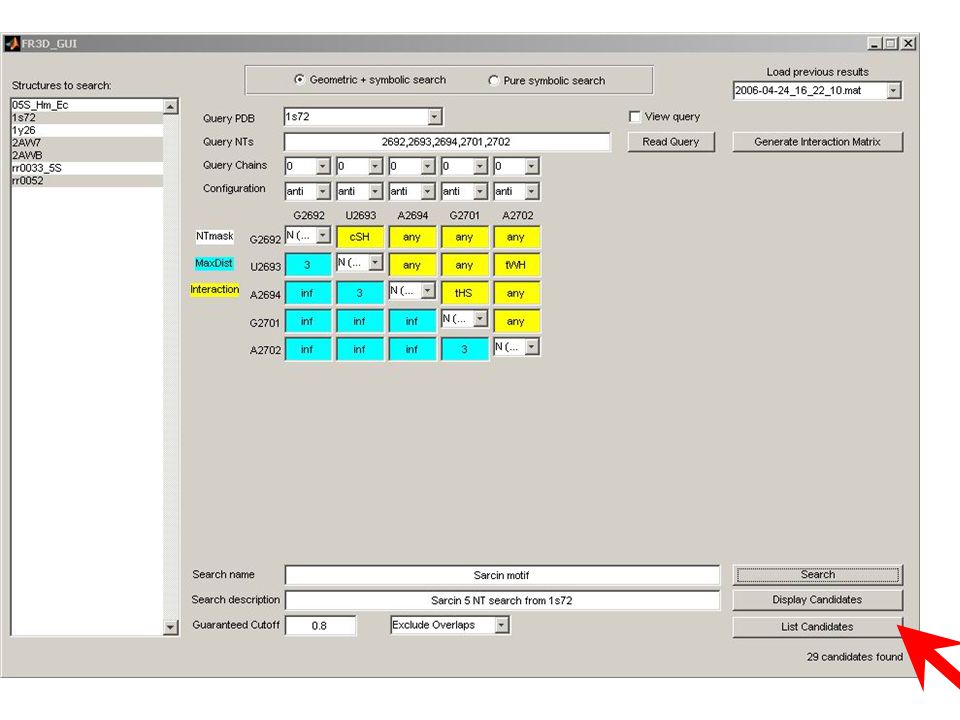
Known Structure Generation Parsing UUAUCCAUGGCGUCGCACAAAGGC CAACAAAAAUAGUUCUGGGAGCAG

8
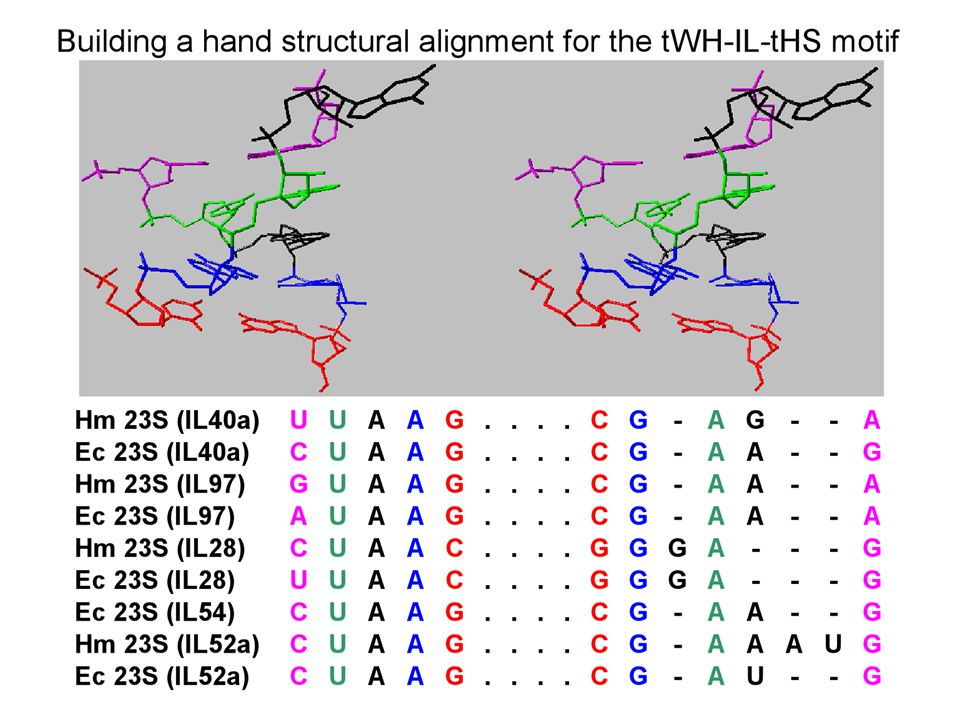
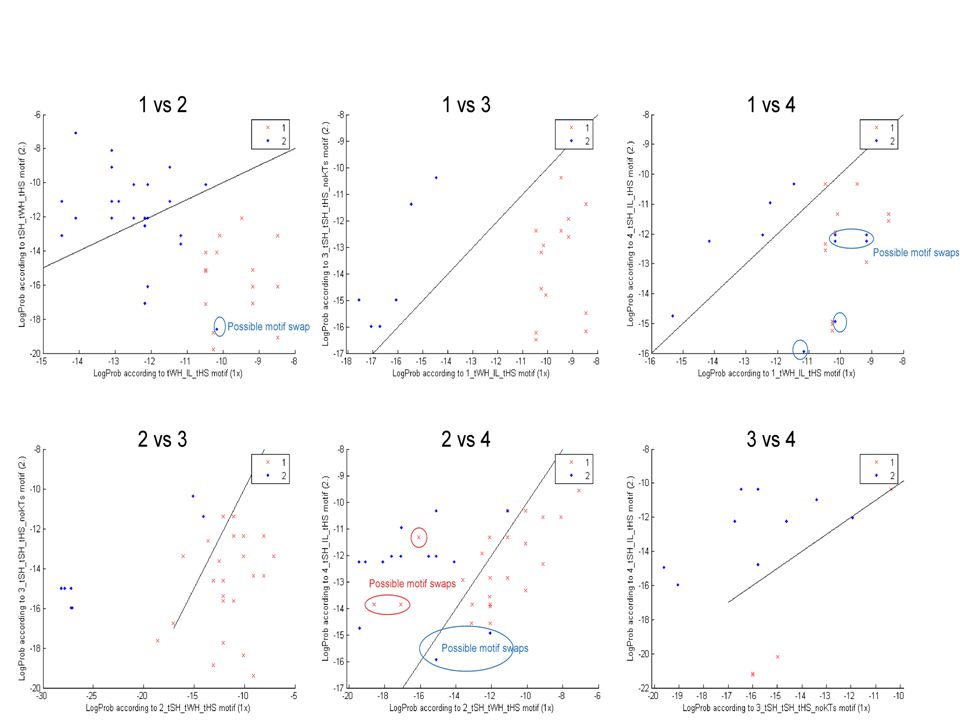
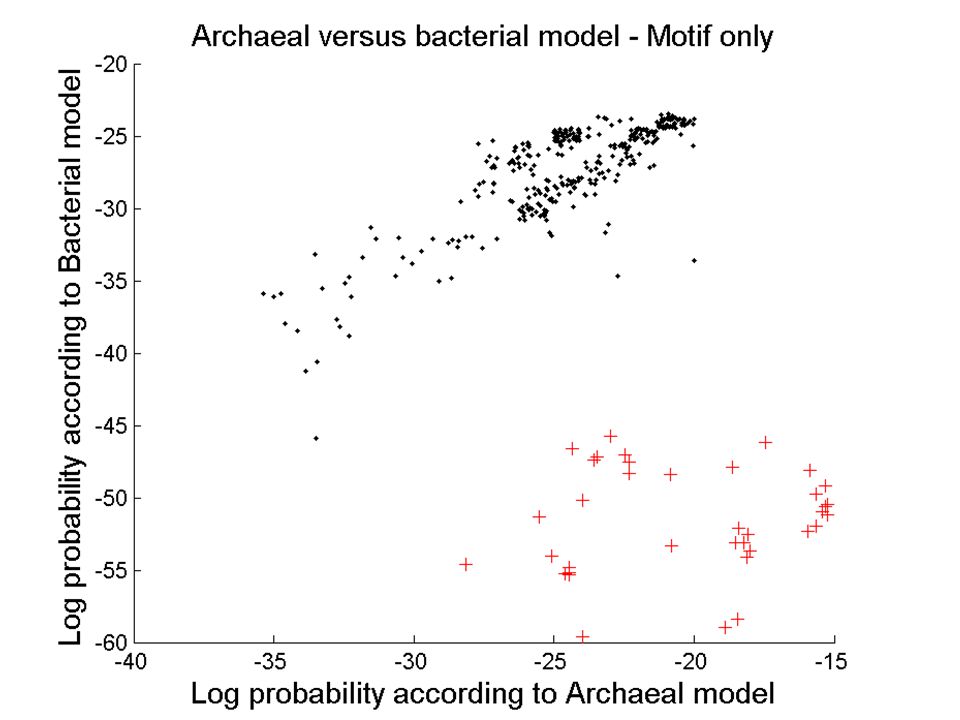
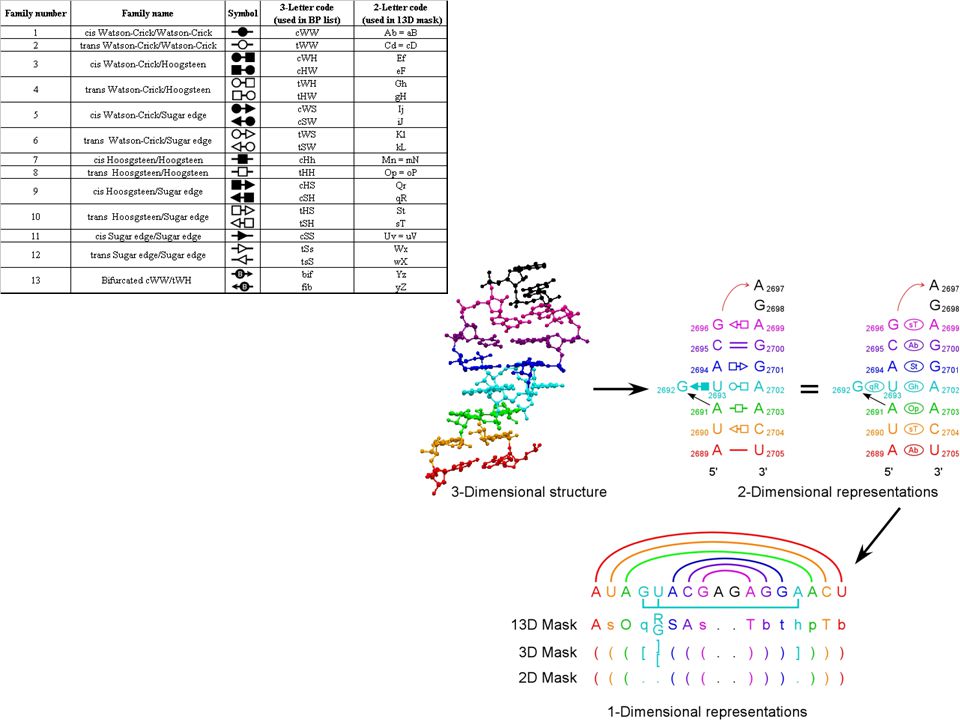
We use SCFG models that are capable of describing not only W.C. interactions, but also all other families of edge- to-edge interactions observed in 3D structures. We program all isosteric subfamilies (figure below) into the SCFG to allow isosteric substitutions when aligning sequences. We also combine SCFG with Markov Random Fields (MRF) models, allowing for the alignment of areas where local crossing interactions occur, or where multiple interactions with a common nucleotide take place. SCFG/MRF are thus capable of generating clusters of bases at once (triples, quadruples, etc.), and are not limited to basepairs. The hybrid SCFG/MRF is capable of detecting areas of motif swaps in the alignments from sequence data alone. Eventually it may be possible to detect structural features of small motifs directly from sequence data. SCFG/MRF models
 into the SCFG to allow isosteric substitutions when aligning sequences. We also combine SCFG with Markov Random Fields (MRF) models, allowing for the alignment of areas where local crossing interactions occur, or where multiple interactions with a common nucleotide take place. SCFG/MRF are thus capable of generating clusters of bases at once (triples, quadruples, etc.), and are not limited to basepairs. The hybrid SCFG/MRF is capable of detecting areas of motif swaps in the alignments from sequence data alone. Eventually it may be possible to detect structural features of small motifs directly from sequence data. SCFG/MRF models.")
11
Programs http://rna.bgsu.edu/FR3D GUI ready, will be posted online within days User manual sometime soon… Appearing soon in J. Math. Biol http://rna.bgsu.edu/ribostral MATLAB and compiled version (PC) available Full manual available Appearing soon in Bioinformatics
 available Full manual available Appearing soon in Bioinformatics.")
18
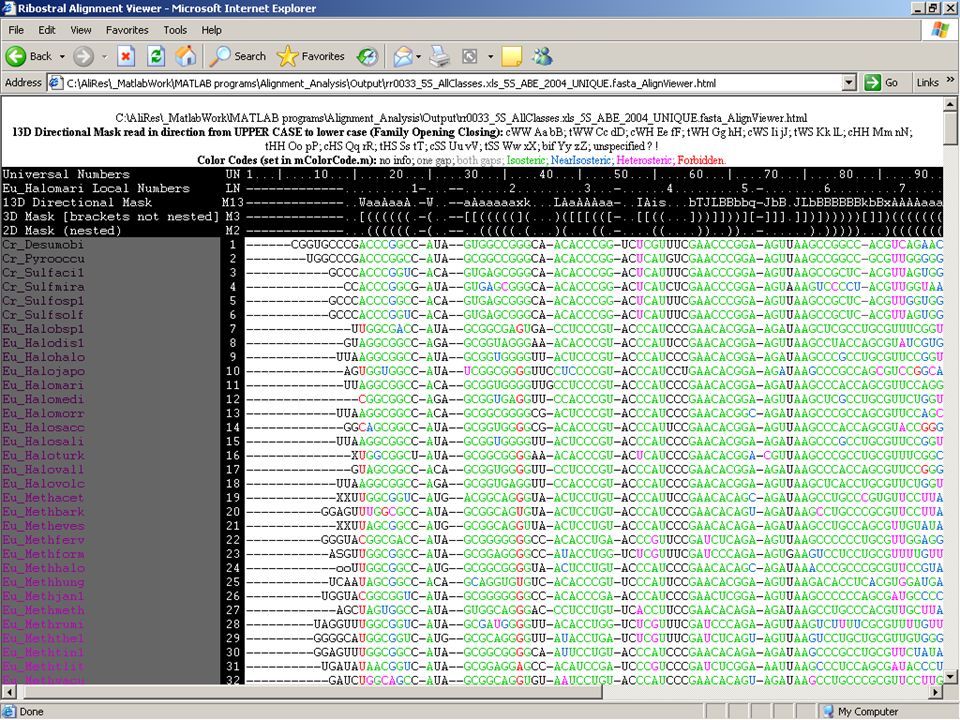
Ribostral Full manual available … Inputs: 1.Fasta alignment file 2.A list of interactions taken from a 3D structure

21
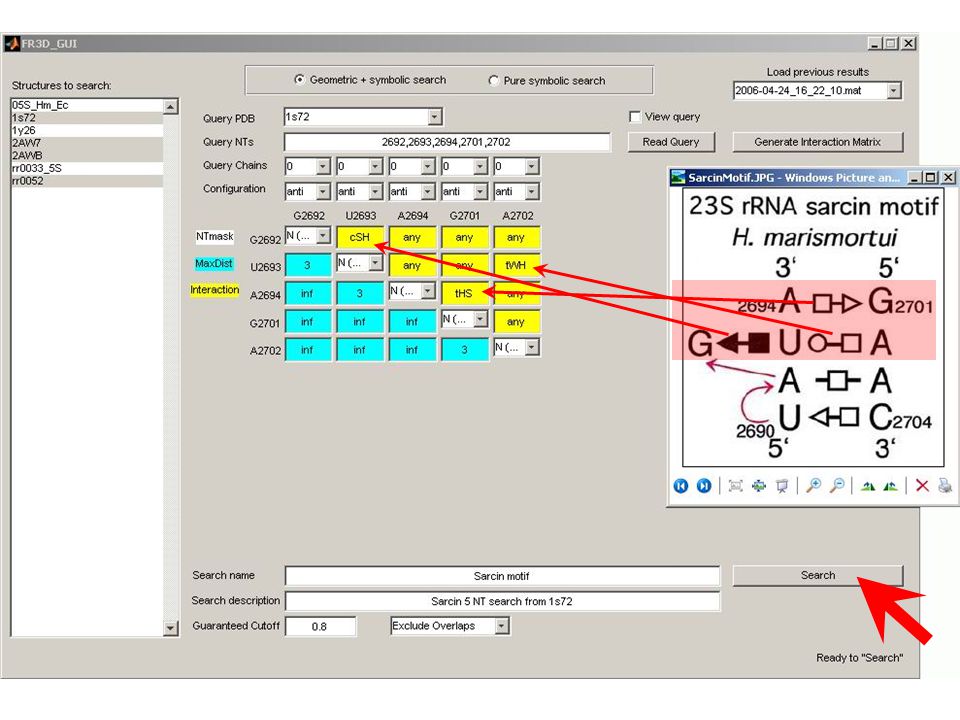
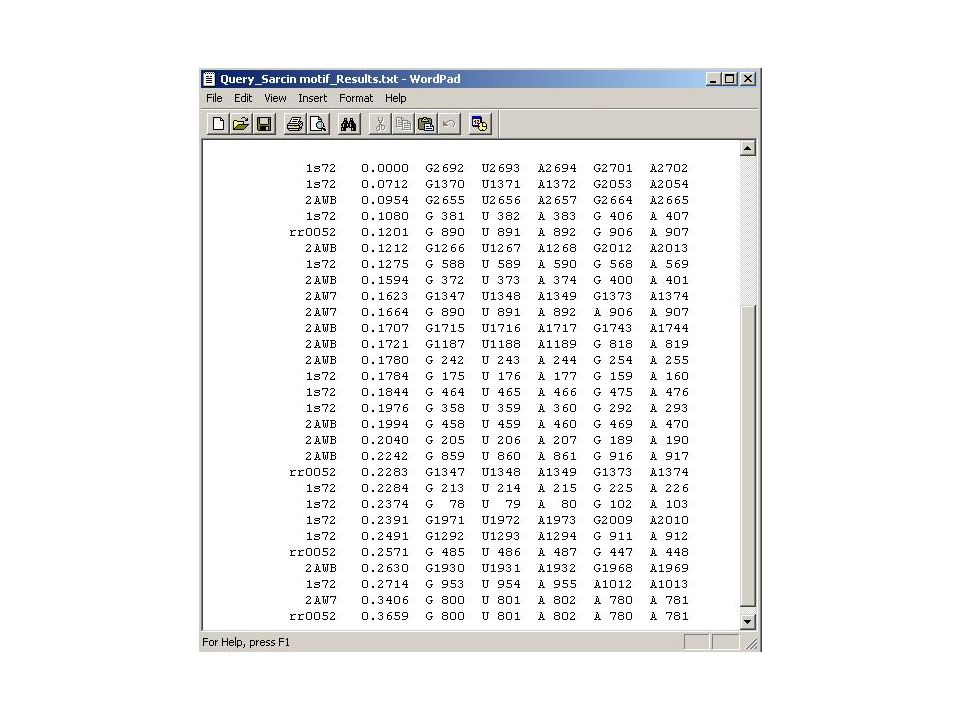
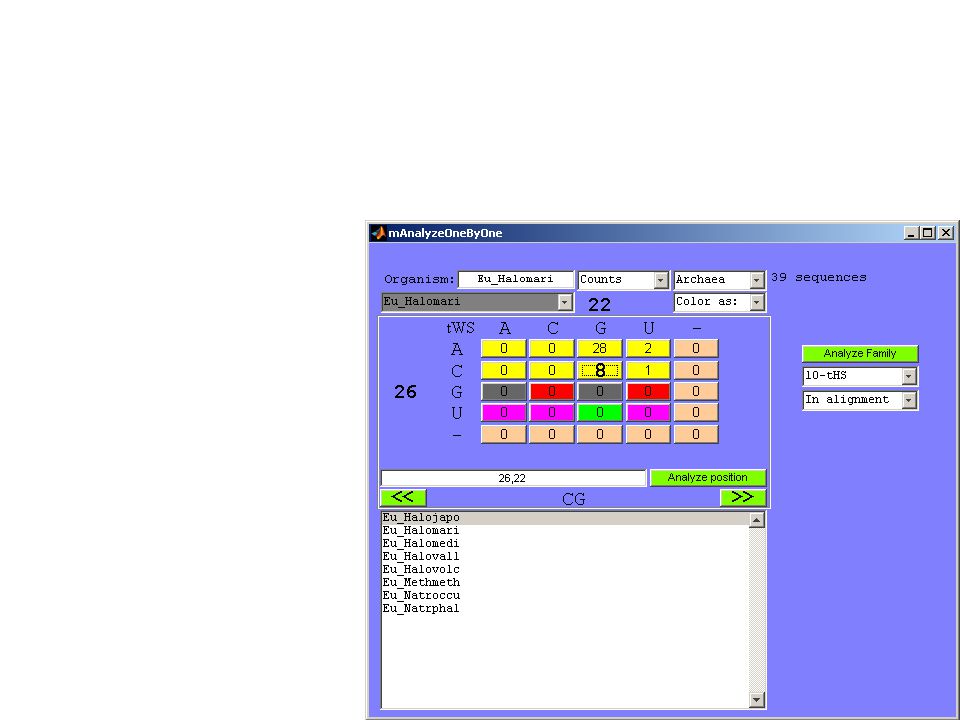
Individual BP score = c x (3I + 2NI – H – 2F – 2G1 – 3G2) Where c is the correction coefficient: c = 100 / (3 x number of sequences) Score calculation: BP 26/22 in Bacteria: 26/22 is tWS CG in the crystal structure. There are: 312 sequences with isosteric (I) substitutions 25 heterosteric (H) substitutions 13 forbidden (F) substitutions Correction coefficient c = 100 / (3x351) = 0.095 Score = 0.095 x (3x312 – 25 – 2x13) = 83
 substitutions 25 heterosteric (H) substitutions 13 forbidden (F) substitutions Correction coefficient c = 100 / (3x351) = Score = x (3x312 – 25 – 2x13) = 83.")
Similar presentations







 for a specific breakout, for a specific round (of breakouts),>")






 Intergenic Repetitive elements Promoters Introns mRNA untranslated region (UTR)>")



![Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]](/16/4993760/big_thumb.jpg "Structure Prediction. Tertiary protein structure: protein folding Three main approaches: [1] experimental determination (X-ray crystallography, NMR) [2]>")



