Download presentation
Presentation is loading. Please wait.

1
The second-simplest cDNA microarray data analysis problem Terry Speed, UC Berkeley Fred Hutchinson Cancer Research Center March 9, 2001

2
cDNA clones (probes) PCR product amplification purification printing microarray Hybridise target to microarray mRNA target) excitation laser 1 laser 2 emission scanning analysis 0.1nl/spot overlay images and normalise
 PCR product amplification purification printing microarray Hybridise target to microarray mRNA target) excitation laser 1 laser 2 emission scanning analysis 0.1nl/spot overlay images and normalise")
3
Biological question Differentially expressed genes Sample class prediction etc. Testing Biological verification and interpretation Microarray experiment Estimation Experimental design Image analysis Normalization Clustering Discrimination R, G 16-bit TIFF files (Rfg, Rbg), (Gfg, Gbg)
, (Gfg, Gbg).")
4
Some motherhood statements Important aspects of a statistical analysis include: Tentatively separating systematic from random sources of variation Removing the former and quantifying the latter, when the system is in control Identifying and dealing with the most relevant source of variation in subsequent analyses Only if this is done can we hope to make more or less valid probability statements

5
The simplest cDNA microarray data analysis problem is identifying differentially expressed genes using one slide This is a common enough hope Efforts are frequently successful It is not hard to do by eye The problem is probably beyond formal statistical inference (valid p-values, etc) for the foreseeable future, and here’s why
 for the foreseeable future, and here’s why")
6
An M vs. A plot M = log 2 (R / G) A = log 2 (R*G) / 2
 A = log 2 (R*G) / 2")
7
Background matters From Spot From GenePix

8
From the NCI60 data set (Stanford web site) No background correction With background correction
 No background correction With background correction")
9
An experiment having within-slide replicates

10
Background makes a difference Background methodSegmentation methodExp1 Exp2 S.nbg66 Gp.nbg76 SA.nbg66 No backgroundQA.fix.nbg76 QA.hist.nbg76 QA.adp.nbg1414 S.valley1721 GP1111 Local surroundingSA1214 QA.fix1823 QA.hist98 QA.adp2726 OthersS.morph99 S.const1414 Medians of the SD of log 2 (R/G) for 8 replicated spots multiplied by 100 and rounded to the nearest integer.
 for 8 replicated spots multiplied by 100 and rounded to the nearest integer.")
11
Normalisation - lowess Global lowess (Matt Callow’s data, LNBL) Assumption: changes roughly symmetric at all intensities.
 Assumption: changes roughly symmetric at all intensities.")
12
From the NCI60 data set (Stanford web site)
")
13
Ngai lab, UCB

14
Tiago’s data from the Goodman lab, UCB

15
From the Ernest Gallo Clinic & Research Center

16
From Peter McCallum Cancer Research Institute, Australia

17
Normalisation - print tip Assumption: For every print group, changes roughly symmetric at all intensities.

18
M vs A after print-tip normalisation

19
Normalization (ctd) Another data set After within slide global lowess normalization. Likely to be a spatial effect. Print-tip groups Log-ratios

20
Assumption: All print-tip-groups have the same spread in M True log ratio is ij where i represents different print-tip-groups and j represents different spots. Observed is M ij, where M ij = a i ij Robust estimate of a i is MAD i = median j { |y ij - median(y ij ) | } Taking scale into account
 | } Taking scale into account.")
21
Normalization (ctd) That same data set Normalization (ctd) That same data set After print-tip location and scale normalization. Incorporate quality measures. Log-ratios Print-tip groups

22
Matt Callow’s Srb1 dataset (#5). Newton’s and Chen’s single slide method
. Newton’s and Chen’s single slide method")
23
Matt Callow’s Srb1 dataset (#8). Newton’s, Sapir & Churchill’s and Chen’s single slide method
. Newton’s, Sapir & Churchill’s and Chen’s single slide method")
24
Genomic DNA vs. Genomic DNA The approach of Roberts et al (Rosetta) Data from Bing Ren
 Data from Bing Ren")
25
The second simplest cDNA microarray data analysis problem is identifying differentially expressed genes using replicated slides There are a number of different aspects: First, between-slide normalization; then What should we look at: averages, SDs t- statistics, other summaries? How should we look at them? Can we make valid probability statements? A report on work in progress

26
Normalization (ctd) Yet another data set Between slides this time (10 here) Only small differences in spread apparent We often see much greater differences Slides Log-ratios
 Yet another data set Between slides this time (10 here) Only small differences in spread apparent We often see much greater differences Slides Log-ratios")
27
Apo A1 Experiments Lowess Normalized M

28
Srb1 Experiments Lowess Normalized M

29
Tiago’s Experiments: mutant fly vs. WT

30
The “NCI 60” experiments (no bg)
")
31
Assumption: All slides have the same spread in M True log ratio is ij where i represents different slides and j represents different spots. Observed is M ij, where M ij = a i ij Robust estimate of a i is MAD i = median j { |y ij - median(y ij ) | } Taking scale into account
 | } Taking scale into account.")
32
Which genes are (relatively) up/down regulated? Two samples. e.g. KO vs. WT or mutant vs. WT TC n n For each gene form the t statistic: average of n trt Ms sqrt(1/n (SD of n trt Ms) 2 ) n n
 2 ) n n.")
33
Which genes are (relatively) up/down regulated? Two samples with a reference (e.g. pooled control) TC* n n For each gene form the t statistic: average of n trt Ms - average of n ctl Ms sqrt(1/n (SD of n trt Ms) 2 + (SD of n ctl Ms) 2 ) C C* n n
 TC* n n For each gene form the t statistic: average of n trt Ms - average of n ctl Ms sqrt(1/n (SD of n trt Ms) 2 + (SD of n ctl Ms) 2 ) C C* n n.")
34
One factor: more than 2 samples Samples: Liver tissue from mice treated by cholesterol modifying drugs. Question 1: Find genes that respond differently between the treatment and the control. Question 2: Find genes that respond similarly across two or more treatments relative to control. T1 C T2T3T4 x 2

35
One factor: more than 2 samples Samples: tissues from different regions of the mouse olfactory bulb. Question 1: differences between different regions. Question 2: identify genes with a pre-specified patterns across regions. T3 T4 T2 T6 T1 T5

36
Two or more factors 6 different experiments at each time point. Dyeswaps. 4 time points (30 minutes, 1 hour, 4 hours, 24 hours) 2 x 2 x 4 factorial experiment. ctlOSM EGF OSM & EGF 4 times
 2 x 2 x 4 factorial experiment. ctlOSM EGF OSM & EGF 4 times.")
37
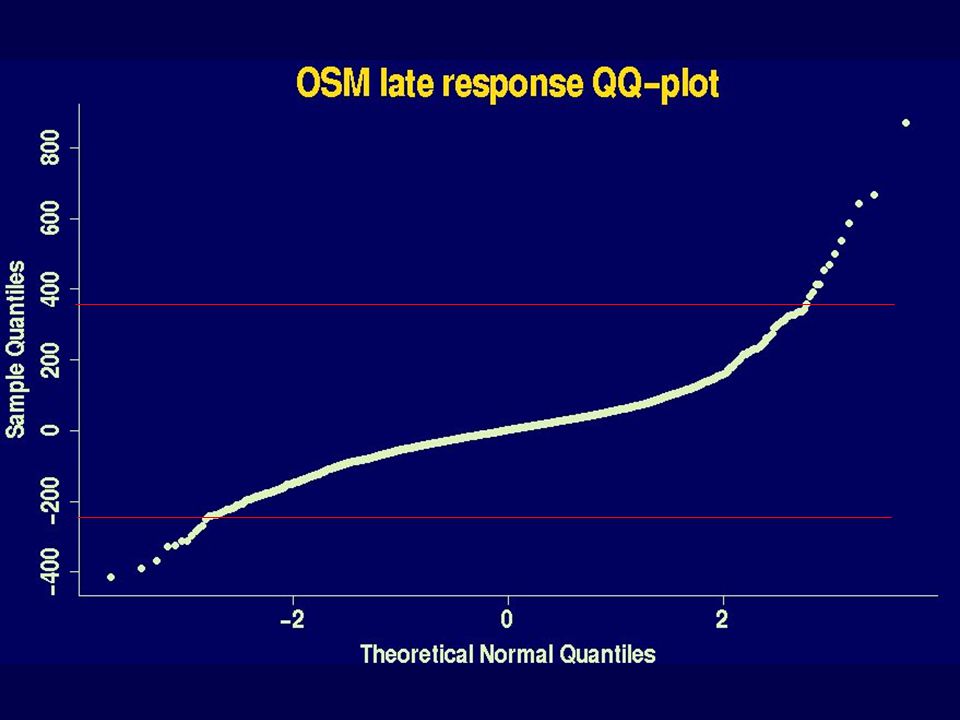
Which genes have changed? When permutation testing possible 1. For each gene and each hybridisation (8 ko + 8 ctl), use M=log 2 (R/G). 2. For each gene form the t statistic: average of 8 ko Ms - average of 8 ctl Ms sqrt(1/8 (SD of 8 ko Ms) 2 + (SD of 8 ctl Ms) 2 ) 3. Form a histogram of 6,000 t values. 4. Do a normal Q-Q plot; look for values “off the line”. 5. Permutation testing. 6. Adjust for multiple testing.
, use M=log 2 (R/G). 2. For each gene form the t statistic: average of 8 ko Ms - average of 8 ctl Ms sqrt(1/8 (SD of 8 ko Ms) 2 + (SD of 8 ctl Ms) 2 ) 3. Form a histogram of 6,000 t values. 4. Do a normal Q-Q plot; look for values off the line . 5. Permutation testing. 6. Adjust for multiple testing..")
38
Histogram & qq plot ApoA1

39
Apo A1: Adjusted and Unadjusted p-values for the 50 genes with the largest absolute t-statistics.

40
Which genes have changed? Permutation testing not possible Our current approach is to use averages, SDs, t-statistics and a new statistic we call B, inspired by empirical Bayes. We hope in due course to calibrate B and use that as our main tool. We begin with the motivation, using data from a study in which each slide was replicated four times.

41
Results from 4 replicates

42
B=LOR compared

43
M t t M Results from the Apo AI ko experiment

44
M t t M Results from the Apo AI ko experiment

45
Empirical Bayes log posterior odds ratio

46
M B t M B t B t M B Results from SR-BI transgenic experiment

47
M B t M B t B t M B Results from SR-BI transgenic experiment

48
Extensions include dealing with Replicates within and between slides Several effects: use a linear model ANOVA: are the effects equal? Time series: selecting genes for trends

49
Un-enriched DNA (Cy3) antibody-enriched DNA (Cy5) Rosetta once more: In vivo Binding Sites of Gal4p in Galactose P <0.001
 antibody-enriched DNA (Cy5) Rosetta once more: In vivo Binding Sites of Gal4p in Galactose P <0.001")
50
Summary (for the second simplest problem) Microarray experiments typically have thousands of genes, but only few (1-10) replicates for each gene. Averages can be driven by outliers. Ts can be driven by tiny variances. B = LOR will, we hope –use information from all the genes –combine the best of M. and T –avoid the problems of M. and T

51
Acknowledgments UCB/WEHI Yee Hwa Yang Sandrine Dudoit Ingrid Lönnstedt Natalie Thorne David Freedman CSIRO Image Analysis Group Michael Buckley Ryan Lagerstorm Ngai lab, UCB Goodman lab, UCB Peter Mac CI, Melb. Ernest Gallo CRC Brown-Botstein lab Matt Callow (LBNL) Bing Ren (WI)
 Bing Ren (WI).")
52
Some web sites: Technical reports, talks, software etc. http://www.stat.berkeley.edu/users/terry/zarray/Html/ Statistical software R “GNU’s S” http://lib.stat.cmu.edu/R/CRAN/ Packages within R environment: -- Spot http://www.cmis.csiro.au/iap/spot.htm -- SMA (statistics for microarray analysis) http://www.stat.berkeley.edu/users/terry/zarray/Software /smacode.html
 /smacode.html.")
53
OSM, EGF and breast cancer Oncostatin M (OSM) is a cytokine in the interleukin 6 (IL-6) family inhibits proliferation of breast caner cells (and other cancer cells) increases the expression of EGRF mRNA Epidermial growth factor (EGF) is a polypeptide growth factor overcomes effects of several breast inhibitors enhances the effect of OSM on breast cancer
 is a cytokine in the interleukin 6 (IL-6) family inhibits proliferation of breast caner cells (and other cancer cells) increases the expression of EGRF mRNA Epidermial growth factor (EGF) is a polypeptide growth factor overcomes effects of several breast inhibitors enhances the effect of OSM on breast cancer")
54
ctl OSM EGF OSM & EGF o e e+oe Factorial experiment design Cell lines Parameters Microarray experiments

55
The microarrays cDNA microarrays were made at PMCI Research Genetics 4 k human gene set + control spots, duplicates =9216 spots 6 different experiments Dyeswaps 4 time points (30 minutes, 1 hour, 4 hours, 24 hours) ~16 spots for each gene in each experiment
 ~16 spots for each gene in each experiment")
56
Back to the factorial design ctl OSM EGF OSM & EGF o e e+oe Different ways of estimating each effect Ex: 1 = ( + o) - ( ) = o 2 - 5 = ( + o + e + oe) - ( ) -(( + o + e + oe)-( + o)) =(o + e + oe) - (e + oe) =o 3 + 6=…=o How do we use all the information? 1 5 3 4 2 6

57
Regression analysis Define a matrix X so that E(M)=X Use least squares estimate for o, e, oe
=X Use least squares estimate for o, e, oe")
59
EGF vs OSM Differentially expressed for EGF or OSM

60
OSM*EGF interaction vs OSM+EGF Differentially expressed for OSM*EGF, OSM or EGF

61
OSM.EGF interaction OSM 10 2 30 minutes EGF OSM.EGF interaction OSM 14 10 EGF OSM.EGF interaction 2 2 3 2 1 OSM 19 3 1 hour 4 hours24 hours 10 OSM 32 EGF OSM.EGF interaction 7 17 2

62
Time series analysis

63
Early and late response genes 1/2 1 4 24 Time M Which genes increase or decrease like the function x 2 ? 1 16 576 1/4 (u)
.")
64
Decompose each graph 1/2 1 4 24 Time True graph x2x2 Left overs = C + D M (u)
")
65
Vector world For each gene, we have a vector, y, of expression estimates at the different time points Project the vector onto the space spanned by the vector u (the values of x 2 at our time points). C is the scalar product u C y u C y

69
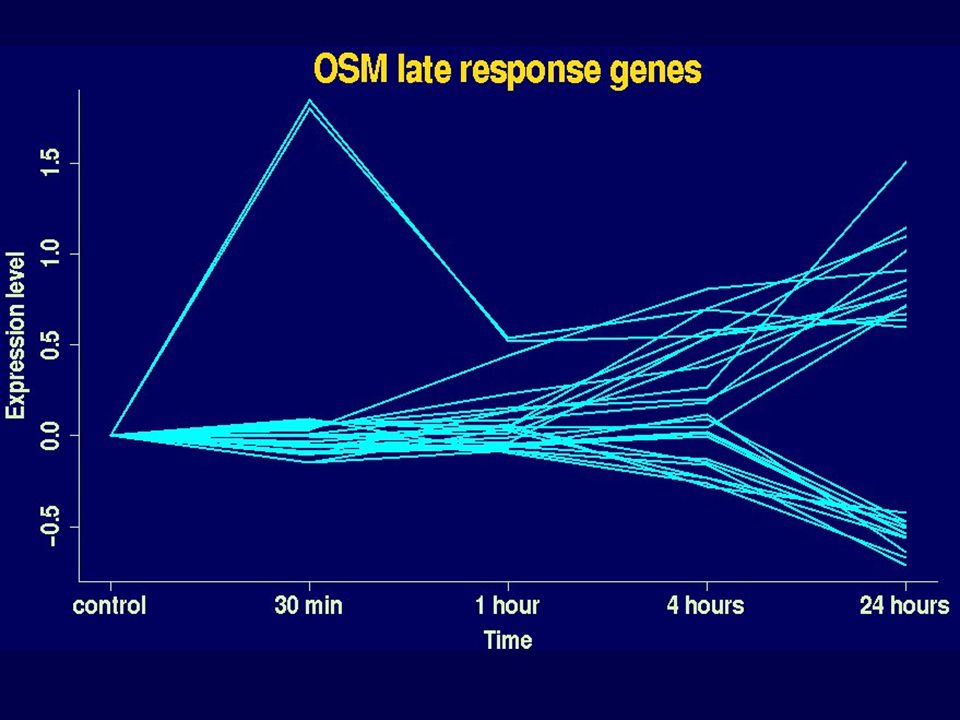
10 OSM EGF OSM.EGF interaction 1 Early response genes 8 OSM EGF OSM.EGF interaction 3 Late response genes 11 2 1 17 25 19 6 1

Similar presentations