Download presentation
Presentation is loading. Please wait.

1
James S.A. Brooke a *, Peter F. Bernath b, Colin M. Western c, Timothy W. Schmidt d, George B. Bacskay d, Marc C. van Hermert e & Gerrit C. Groenenboom f a: Department of Chemistry, University of York, York, UK. b: Department of Chemistry & Biochemistry, Old Dominion University, Norfolk, VA, USA. c: School of Chemistry, University of Bristol, Bristol, UK. d: School of Chemistry, The University of Sydney, New South Wales, Australia. e: Department of Chemistry, Gorlaeus Laboratories, Leiden University, The Netherlands. f: Theoretical Chemistry, Institute for Molecules and Materials (IMM), Radboud University Nijmegen, Nijmegen, The Netherlands. Funded by:
, Radboud University Nijmegen, Nijmegen, The Netherlands. Funded by:.")
2
Line lists Quantum number assignments, line positions and intensities Positions – Recorded directly from laboratory spectra Intensities – Obtained with a combination of experimental and theoretical methods – Require potential energy curve and (transition) dipole moment function
 dipole moment function")
3
Potential Energy Curves – Experimental (1) Spectrum is obtained from lab observations Lines are assigned and a fit of line positions provides molecular constants for each vibrational level. RKR procedure generates potential energy curve – Bob Le Roy’s RKR1 program Spectrum Molecular constants Line assignment and fit RKR1 Potential energy curve fit Equilibrium constants

4
Potential Energy Curves – Experimental (2) C 2 Swan system (d 3 Π g -a 3 Π u ) potential energy curves and TDM function d
 C 2 Swan system (d 3 Π g -a 3 Π u ) potential energy curves and TDM function d")
5
(Transition) Dipole Moment Function - Theoretical Line intensities cannot be accurately obtained from the experimental spectra that we use. Obtained from ab initio methods Usually using good level of theory, e.g. MRCI, and basis set such as aug-cc-PV6Z (used for NH), as they’re only diatomic molecules Ab initio Electronic (transition) dipole moment function
, as they’re only diatomic molecules Ab initio Electronic (transition) dipole moment function.")
6
LEVEL and PGOPHER to Final Line List Bob Le Roy’s LEVEL calculates vibrational wavefunctions by solving the one- dimensional Schrödinger equation. Vibrational wavefunctions and transition dipole moment matrix elements LEVEL Potential energy curve + Electronic (transition) dipole moment function
 dipole moment function.")
7
Transition Dipole Moment Matrix Elements It then overlaps them and multiplies the result by the electronic (transition) dipole moment function, which is then integrated to give the transition dipole moment matrix element (TDMME).
 dipole moment function, which is then integrated to give the transition dipole moment matrix element (TDMME).")
8
LEVEL and PGOPHER to Final Line List Bob Le Roy’s LEVEL calculates vibrational wavefunctions by solving the one-dimensional Schrödinger equation. Vibrational wavefunctions and transition dipole moment matrix elements Einstein As and f-values – line list with positions and intensities LEVEL PGOPHER Potential energy curve + Electronic (transition) dipole moment function It then overlaps them and multiplies the result by the electronic (transition) dipole moment function, which is then integrated to give the TDMME. Colin Western’s PGOPHER calculates Hönl-London factors, and combines them with the TDMMEs and line positions to calculate Einstein A values and then f-values.
 dipole moment function It then overlaps them and multiplies the result by the electronic (transition) dipole moment function, which is then integrated to give the TDMME. Colin Western’s PGOPHER calculates Hönl-London factors, and combines them with the TDMMEs and line positions to calculate Einstein A values and then f-values..")
9
Line Strength Equations PGOPHER calculates Hönl-London factors, and combines them with the TDMMEs and line positions to calculate Einstein A values:

10
LEVEL and PGOPHER to Final Line List Bob Le Roy’s LEVEL calculates vibrational wavefunctions by solving the one-dimensional Schrödinger equation. Vibrational wavefunctions and transition dipole moment matrix elements Einstein As and f-values – line list with positions and intensities LEVEL PGOPHER Potential energy curve + Electronic (transition) dipole moment function It then overlaps them and multiplies the result by the electronic (transition) dipole moment function, which is then integrated to give the TDMME. Colin Western’s PGOPHER calculates Hönl-London factors, and combines them with the TDMMEs and line positions to calculate Einstein A values and then f-values. A final line list is created including positions and intensities for observed and non-observed transitions.
 dipole moment function It then overlaps them and multiplies the result by the electronic (transition) dipole moment function, which is then integrated to give the TDMME. Colin Western’s PGOPHER calculates Hönl-London factors, and combines them with the TDMMEs and line positions to calculate Einstein A values and then f-values. A final line list is created including positions and intensities for observed and non-observed transitions..")
11
Line strengths Ab initio Electronic (transition) dipole moment function Vibrational wavefunctions and transition dipole moment matrix elements Einstein As and f-values – line list with positions and intensities + LEVEL Start PGOPHER Spectrum Molecular constants Line assignment and fit RKR1 Potential energy curve fit Equilibrium constants End
 dipole moment function Vibrational wavefunctions and transition dipole moment matrix elements Einstein As and f-values – line list with positions and intensities + LEVEL Start PGOPHER Spectrum Molecular constants Line assignment and fit RKR1 Potential energy curve fit Equilibrium constants End")
12
C 2 Swan System - Introduction C 2 is found in interstellar clouds, comets, cool stars and flames. Available line strengths based on assignments from 1968 by Phillips and Davis [1] Tanabashi and Amano in 2002 [2] disagreed with Phillips and Davis. Tanabashi et al. in 2007 [3] confirmed the disagreement. A new line list would be very beneficial. [1] Phillips JG, Davis SP. The Swan system of the C 2 molecule, The spectrum of the HgH molecule. University of California Press; 1968. [2] Tanabashi A, Amano T. J Mol Spectrosc 2002;215:285–94. [3] Tanabashi A, Hirao T, Amano T, Bernath PF. Astrophys J Suppl Ser 2007;169:472–84. Swan system is the most prominent and therefore heavily studied

13
C 2 Swan System – Perturbations (1) Most vibrational levels of the d 3 Π g state are perturbed. Worst affected are the v=4 and 6 levels. Perturbations quantified by Bornhauser et al. in 2010 and 2011 [4,5] – a 5 Π g state also discovered and identified to perturb d 3 Π g v=6 We refitted the available line positions and included these perturbations, and new data from Yeung et al.. [4] Bornhauser P, Knopp G, Gerber T, Radi P. J Mol Spectrosc 2010;262:69–74. http://dx.doi.org/10.1016/j.jms.2010.05.008. [5] Bornhauser P, Sych Y, Knopp G, Gerber T, Radi PP. J Chem Phys 2011;134:044302. http://dx.doi.org/10.1063/1.3526747. [6] Yeung SH, Chan MC, Wang N, Cheung. Chem Phys Lett 2013;557:31–6 doi: http://dx.doi.org/10.1016/j.cplett.2012.11.092.http://dx.doi.org/10.1016/j.cplett.2012.11.092 u

14
C 2 Swan System – Perturbations (2) Change in perturbation constants in fit: ParameterBornhauser et al. valueValue from fit -0.6401(86)-0.6147(59) 0.24737(61)0.24869(21) 0.7855(110)0.7417(82) 0.31192(37)0.31123(12) 4.6220(88)4.6150(94) Change in line errors with inclusion of perturbation constants: Involving upper vibrational level Average line position error (cm -1 ) Without perturbation constants With perturbation constants 40.2100.069 60.5710.038 all0.0710.025
 (59) (61) (21) (110)0.7417(82) (37) (12) (88)4.6150(94) Change in line errors with inclusion of perturbation constants: Involving upper vibrational level Average line position error (cm -1 ) Without perturbation constants With perturbation constants all")
15
C 2 Swan System – Intensities Electronic transition dipole moment calculated by Tim Schmidt and George Bacskay (University of Sydney) [8] Earlier mentioned procedure performed v’ Our value (ns) Theor. [8] (ns) Expt. (ns) Expt. (ns) 98.095.1101.8 ± 4.2106 ± 15 99.896.7 96.7 ± 5.2 105 ± 15 2 102.499.1 104.0 ± 17 106.0102 110.9107 5 118.2113 Vibrational lifetime comparison [8] Schmidt TW, Bacskay GB. J Chem Phys 2007;127(23).
![C 2 Swan System – Intensities Electronic transition dipole moment calculated by Tim Schmidt and George Bacskay (University of Sydney) [8] Earlier mentioned procedure performed v’ Our value (ns) Theor.](http://images.slideplayer.com/15/4756574/slides/slide_15.jpg "[8] (ns) Expt. (ns) Expt. (ns) ± ± ± ± ± Vibrational lifetime comparison [8] Schmidt TW, Bacskay GB. J Chem Phys 2007;127(23)..")
16
C 2 Swan System – Data Included Final line list with positions and intensities produced: v' v'' J' J'' F' F'' p' p'' Observed Calculated Residual Perturbation E'' A f Description Paper Quality Weight 0 0 2 3 1 1 e e 19371.76200 19371.7621 -0.00010 0.00000 -1.4481 2.621927E+6 7.481903E-3 pP1e(3) T7 a 0.00500 0 0 3 4 1 1 f f 19369.31700 19369.3187 -0.00170 0.00000 9.8325 3.363432E+6 1.045363E-2 pP1f(4) T7 a 0.00500 0 0 4 5 1 1 e e 19367.02600 19367.0221 0.00390 0.00000 24.0522 3.652000E+6 1.194297E-2 pP1e(5) T7 a 0.00500 0 0 5 6 1 1 f f 19364.84600 19364.8809 -0.03490 0.00000 41.3152 3.780763E+6 1.278964E-2 pP1f(6) T7 c 0.05000 0 0 6 7 1 1 e e 19362.97400 19362.9435 0.03050 0.00000 61.5972 3.844201E+6 1.332207E-2 pP1e(7) T7 c 0.05000 0 0 7 8 1 1 f f 19361.19500 19361.1967 -0.00170 0.00000 85.0354 3.874655E+6 1.367316E-2 pP1f(8) T7 a 0.00500 0 0 8 9 1 1 e e 19359.69200 19359.6924 -0.00040 0.00000 111.5372 3.890445E+6 1.392373E-2 pP1e(9) T7 a 0.00500 0 0 9 10 1 1 f f 19358.39500 19358.3892 0.00580 0.00000 141.2663 3.896652E+6 1.410409E-2 pP1f(10) T7 a 0.00500 0 0 10 11 1 1 e e 19357.36400 19357.3560 0.00800 0.00000 174.0863 3.899270E+6 1.424427E-2 pP1e(11) T7 a 0.00500 0 0 11 12 1 1 f f 19356.51400 19356.5168 -0.00280 0.00000 210.1675 3.898503E+6 1.435123E-2 pP1f(12) T7 a 0.00500 0 0 12 13 1 1 e e 19355.95800 19355.9729 -0.01490 0.00000 249.3643 3.897172E+6 1.443957E-2 pP1e(13) T7 a 0.00500 0 0 13 14 1 1 f f 19355.61300 19355.6049 0.00810 0.00000 291.8268 3.894456E+6 1.450963E-2 pP1f(14) T7 a 0.00500 0 0 14 15 1 1 e e 19355.61300 19355.5592 0.05380 0.00000 337.4353 3.892098E+6 1.457015E-2 pP1e(15) T7 y 99999.00000 0 0 15 16 1 1 f f 19355.61300 19355.6644 -0.05140 0.00000 386.2900 3.889092E+6 1.461975E-2 pP1f(16) T7 c 0.05000 0 0 16 17 1 1 e e 19356.15900 19356.1219 0.03710 0.00000 438.3304 3.886728E+6 1.466390E-2 pP1e(17) T7 y 99999.00000 0 0 17 18 1 1 f f 19356.69700 19356.6999 -0.00290 0.00000 493.5777 3.884009E+6 1.470083E-2 pP1f(18) T7 a 0.00500 0 0 18 19 1 1 e e 19357.64500 19357.6639 -0.01890 0.00000 552.0606 3.882009E+6 1.473487E-2 pP1e(19) T7 c 0.05000 0 0 19 20 1 1 f f 19358.71500 19358.7135 0.00150 0.00000 613.6943 3.879775E+6 1.476359E-2 pP1f(20) T7 a 0.00500 0 0 20 21 1 1 e e 19360.19400 19360.1861 0.00790 0.00000 678.6244 3.878267E+6 1.479079E-2 pP1e(21) T7 a 0.00500 0 0 21 22 1 1 f f 19361.70600 19361.7057 0.00030 0.00000 746.6338 3.876572E+6 1.481407E-2 pP1f(22) T7 a 0.00500 Published in JQSRT [9]. Line list available from article website, http://uk.arxiv.org/abs/1212.2102, and http://bernath.uwaterloo.ca/download/AutoIndex.php?dir=/C2/ [9] Brooke JSA, Bernath PF, Schmidt TW, Bacskay GB. JQSRT 2013;124(0):11-20. doi:10.1016/j.jqsrt.2013.02.025.
![C 2 Swan System – Data Included Final line list with positions and intensities produced: v v J J F F p p Observed Calculated Residual Perturbation E A f Description Paper Quality Weight e e E E-3 pP1e(3) T7 a f f E E-2 pP1f(4) T7 a e e E E-2 pP1e(5) T7 a f f E E-2 pP1f(6) T7 c e e E E-2 pP1e(7) T7 c f f E E-2 pP1f(8) T7 a e e E E-2 pP1e(9) T7 a f f E E-2 pP1f(10) T7 a e e E E-2 pP1e(11) T7 a f f E E-2 pP1f(12) T7 a e e E E-2 pP1e(13) T7 a f f E E-2 pP1f(14) T7 a e e E E-2 pP1e(15) T7 y f f E E-2 pP1f(16) T7 c e e E E-2 pP1e(17) T7 y f f E E-2 pP1f(18) T7 a e e E E-2 pP1e(19) T7 c f f E E-2 pP1f(20) T7 a e e E E-2 pP1e(21) T7 a f f E E-2 pP1f(22) T7 a Published in JQSRT [9].](http://images.slideplayer.com/15/4756574/slides/slide_16.jpg "Line list available from article website, and dir=/C2/ [9] Brooke JSA, Bernath PF, Schmidt TW, Bacskay GB. JQSRT 2013;124(0): doi: /j.jqsrt")
17
[10] Boudjaadar D, Brion J, Chollet P, Guelachvili G, Vervloet M. J Mol Spectrosc 1986;119(2):352-66. doi:10.1016/0022-2852(86)90030-5. [11] Ram RS, Bernath PF, Hinkle KH. J Chem Phys 1999;110:5557-63. doi:10.1063/1.478453. [12] Ram RS, Bernath PF. J Mol Spectrosc 2010;260:115-9. doi:10.1016/j.jms.2010.01.006. [13] Robinson A, Brown J, Flores-Mijangos J, Zink L, Jackson M. Mol Phys 2007;105:639-62. doi:10.1080/00268970601162085.
![[10] Boudjaadar D, Brion J, Chollet P, Guelachvili G, Vervloet M.](http://images.slideplayer.com/15/4756574/slides/slide_17.jpg "J Mol Spectrosc 1986;119(2): doi: / (86) [11] Ram RS, Bernath PF, Hinkle KH. J Chem Phys 1999;110: doi: / [12] Ram RS, Bernath PF. J Mol Spectrosc 2010;260: doi: /j.jms [13] Robinson A, Brown J, Flores-Mijangos J, Zink L, Jackson M. Mol Phys 2007;105: doi: /")
18
No new line position fit - used Ram and Bernath 2010 [12] Mostly same procedure as before Dipole moment function calculated by Gerrit Groenenboom – not published [14] [14] Campbell WC, Groenenboom GC, Lu HI, Tsikata E, Doyle JM. Phys Rev Lett 2008;100(8). doi:10.1103/PhysRevLett.100.083003.
![No new line position fit - used Ram and Bernath 2010 [12] Mostly same procedure as before Dipole moment function calculated by Gerrit Groenenboom – not published [14] [14] Campbell WC, Groenenboom GC, Lu HI, Tsikata E, Doyle JM.](http://images.slideplayer.com/15/4756574/slides/slide_18.jpg "Phys Rev Lett 2008;100(8). doi: /PhysRevLett")
19
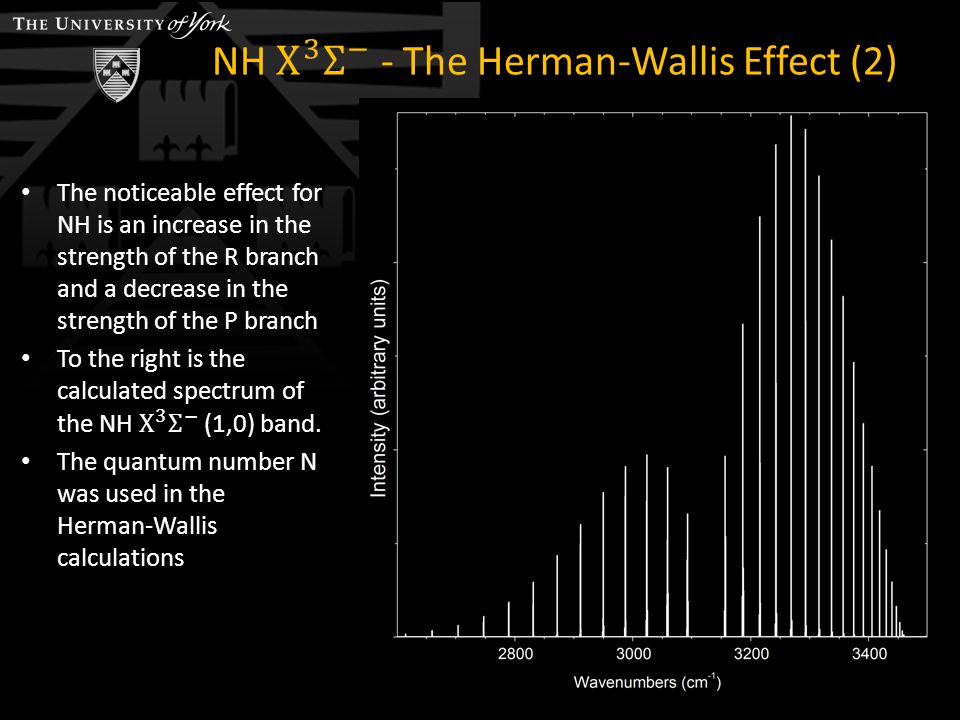
Rotation - centrifugal force - causes change in bond length Results in a change in the vibrational wavefunctions TDMMEs are changed Effect is greater in NH than C 2 due to light H atom

22
CN A 2 П-X 2 Σ + and B 2 Σ + -X 2 Σ + systems and the CP A 2 П-X 2 Σ + system Will be discussed tomorrow by Ram Herman-Wallis effect is not as strong in these systems as the atoms are heavier Maximum Herman-Wallis effect for observed bands is: – 70% for CN A 2 П-X 2 Σ + – 40% for CN B 2 Σ + -X 2 Σ + – 2% for CP A 2 П-X 2 Σ + (up to 30% for unobserved bands)
")
23
Conclusions New line list with positions and intensities produced for C 2 Swan system – up to v′=10 and v′=9. Similar lists will soon be finished for NH, CN and CP. The combination of experimental and theoretical results is very effective and will be used for more molecules in the future. The Herman-Wallis effect is very important for NH, as expected, but also affects heavier diatomic molecules. Thanks for listening!

24
Standard definitionUsed for NH

25
Possible values of ΔN 6 5 7 5 4 6 4 3 5 J N 6 5 7 5 4 6 4 3 5 6 5 4 6 5 4 7 6 8 3 2 4 7 3 ΔJΔJ ΔNΔN 0+1+1 0+10 0+1+1 ΔJΔJ ΔNΔN ΔJΔJ ΔNΔN +1 -3 +1 +1 +3

Similar presentations






 using a quantum cascade laser- based cavity ringdown.>")













