Download presentation
Presentation is loading. Please wait.

1
Approach to myopathy

2
The first goal in approaching a patient with a suspected muscle disease is to determine the correct site of the lesion. The second goal is to determine the cause of the myopathy. The third goal is to determine whether a specific treatment is available and if not, to optimally manage the patient’s symptoms to maximize his or her functional abilities and enhance quality of life.

4
History Clinical evaluation Investigations Examination

5
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

6
Which negative and/or positive symptoms and signs does the patient demonstrate?
Weakness Myalgia Fatigue Cramps Exercise intolerance Contractures Muscle atrophy myotonia myoglobinuria

7
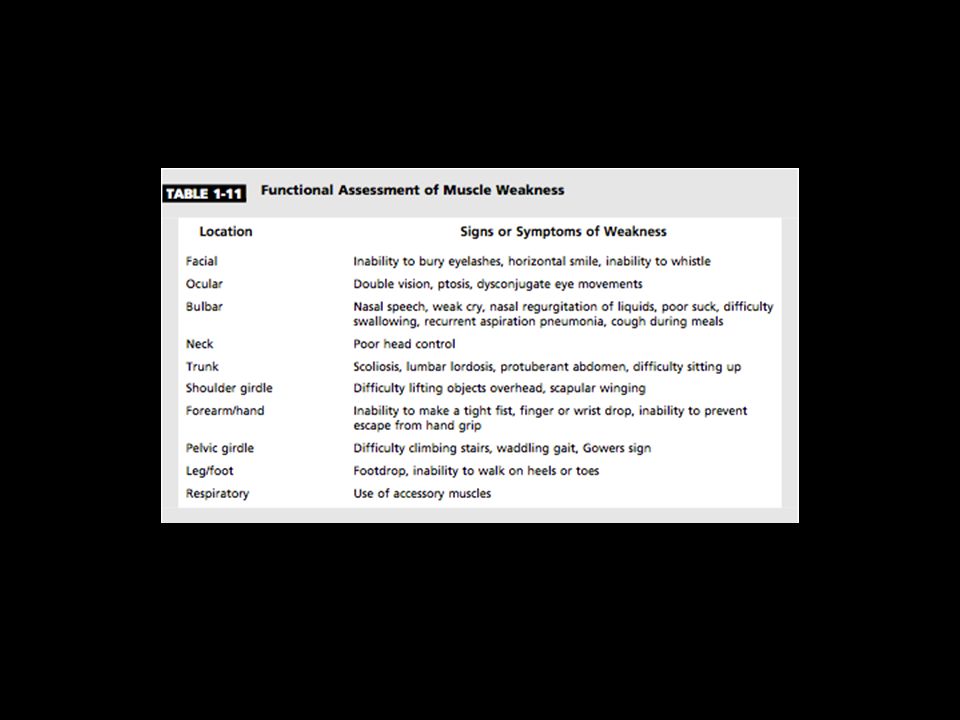
Weakness: Proximal lower extremities: Proximal upper extremities:
difficulty climbing stairs, arising from a low chair or toilet, or getting up from a squatted position. Proximal upper extremities: trouble lifting objects over their head and brushing their hair. Distal upper extremities: difficulty opening jars, inability to turn a key in the ignition. Distal lower extremities: tripping due to footdrop. Cranial muscle weakness, dysarthria, dysphagia, or ptosis.

8
Fatigue less useful negative symptom Nonspecific
abnormal fatigability after exercise can result from certain metabolic and mitochondrial myopathies, and it is important to define the duration and intensity of exercise that provokes the fatigue.

9
Myalgia, like fatigue, is another nonspecific symptom
episodic (metabolic myopathies) constant (inflammatory muscle disorders) more likely to be due to orthopedic or rheumatological disorders
 constant (inflammatory muscle disorders) more likely to be due to orthopedic or rheumatological disorders.")
10
Muscle cramp They are typically benign, occurring frequently in normal individuals, and are seldom a feature of a primary myopathy Cramps can occur with: dehydration, hyponatremia, azotemia, myxedema disorders of the nerve or motor neuron (especially amyotrophic lateral sclerosis).
.")
11
Muscle contractures uncommon
They are typically provoked by exercise in patients with glycolytic enzyme defects. Contractures differ from cramps in that they usually last longer and are electrically silent with needle EMG. EDMD also cause contractures

12
Myotonia is the phenomenon of impaired relaxation of muscle after forceful voluntary contraction and most commonly involves the hands and eyelids. Myotonia is due to repetitive depolarization of the muscle membrane. Patients may complain of muscle stiffness or tightness resulting in difficulty releasing their handgrip after a handshake, unscrewing a bottle top, or opening their eyelids if they forcefully shut their eyes. Myotonia classically improves with repeated exercise.

13
paramyotonia congenita: ‘‘paradoxical myotonia’’ in that symptoms are typically worsened by exercise or repeated muscle contractions. Exposure to cold results in worsening of both myotonia and paramyotonia.

14
Myopathies associated with muscle stiffness:
Myotonic dystrophy Proxymal myotonic dystrophy Myotonia congenita Paramyotonia congenita Hyperkalemic periodic paralysis Hypothyroid myopathy

15
Myoglobinuria uncommon manifestation of muscle disease
caused by the excessive release of myoglobin from muscle during periods of rapid muscle destruction (rhabdomyolysis). Severe myoglobinuria can result in renal failure due to acute tubular necrosis. If patients complain of exercise-induced weakness and myalgias, they should be asked if their urine has ever turned cola-colored or red during or after these episodes. Recurrent myoglobinuria is usually due to an underlying metabolic myopathy isolated episodes, particularly occurring after unaccustomed strenuous exercise, are frequently idiopathic.
. Severe myoglobinuria can result in renal failure due to acute tubular necrosis. If patients complain of exercise-induced weakness and myalgias, they should be asked if their urine has ever turned cola-colored or red during or after these episodes. Recurrent myoglobinuria is usually due to an underlying metabolic myopathy. isolated episodes, particularly occurring after unaccustomed strenuous exercise, are frequently idiopathic.")
17
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

18
What is the temporal evolution?
onset, duration, and evolution of the patient’s symptoms

19
Myopathies Presenting at Birth
Congenital myotonic dystrophy Centronuclear (myotubular) myopathy Congenital fiber-type disproportion Central core disease Nemaline (rod) myopathy Congenital muscular dystrophy Lipid storage diseases (carnitine deficiency) Glycogen storage diseases (acid maltase and phosphorylase deficiencies)
 myopathy. Congenital fiber-type disproportion. Central core disease. Nemaline (rod) myopathy. Congenital muscular dystrophy. Lipid storage diseases (carnitine deficiency) Glycogen storage diseases (acid maltase and phosphorylase deficiencies)")
20
Myopathies Presenting in Childhood
Muscular dystrophies Lipid storage disease (carnitine deficiency) Duchenne Becker Glycogen storage disease (acid maltase deficiency) Emery-Dreifuss Facioscapulohumeral Limb-girdle Mitochondrial myopathies Congenital Inflammatory myopathies Endocrine-metabolic disorders Dermatomyositis Polymyositis (rarely) Hypokalemia Congenital myopathies Hypocalcemia Hypercalcemia Nemaline Centronuclear Central core
 Duchenne. Becker. Glycogen storage disease (acid maltase deficiency) Emery-Dreifuss. Facioscapulohumeral. Limb-girdle. Mitochondrial myopathies. Congenital. Inflammatory myopathies. Endocrine-metabolic disorders. Dermatomyositis. Polymyositis (rarely) Hypokalemia. Congenital myopathies. Hypocalcemia. Hypercalcemia. Nemaline. Centronuclear. Central core.")
21
Myopathies Presenting in Adulthood
Muscular dystrophies Thyroid Parathyroid Limb-girdle Adrenal Facioscapulohumeral Pituitary disorders Becker Toxic myopathies Emery-Dreifuss Inflammatory myopathies Alcohol Corticosteroids Polymyositis Local injections of narcotics Dermatomyositis Colchicine Inclusion body myositis Chloroquine Viral [HIV] Statins Metabolic myopathies Myotonic dystrophy Acid maltase deficiency Distal myopathies Lipid storage diseases Debrancher deficiency Nemaline myopathy Centronuclear myopathy Phosphorylase b kinase deficiency Mitochondrial myopathies Endocrine myopathies Identifying the age that symptoms began can provide crucial information leading to the correct diagnosis. For example, symptoms of Duchenne mus- cular dystrophy usually are identified byage 3, whereas most facioscapulohum- eral and limb-girdle muscular dystro- phies (LGMDs) begin in adolescence or later. Of the inflammatory myopathies, dermatomyositis occurs in children andadults; polymyositis occurs rarely in children but at any decade in the adult years; and inclusion body myositis oc- curs most commonly in the elderly.
 begin in adolescence or later. Of the inflammatory myopathies, dermatomyositis occurs in children andadults; polymyositis occurs rarely in children but at any decade in the adult years; and inclusion body myositis oc- curs most commonly in the elderly.")
22
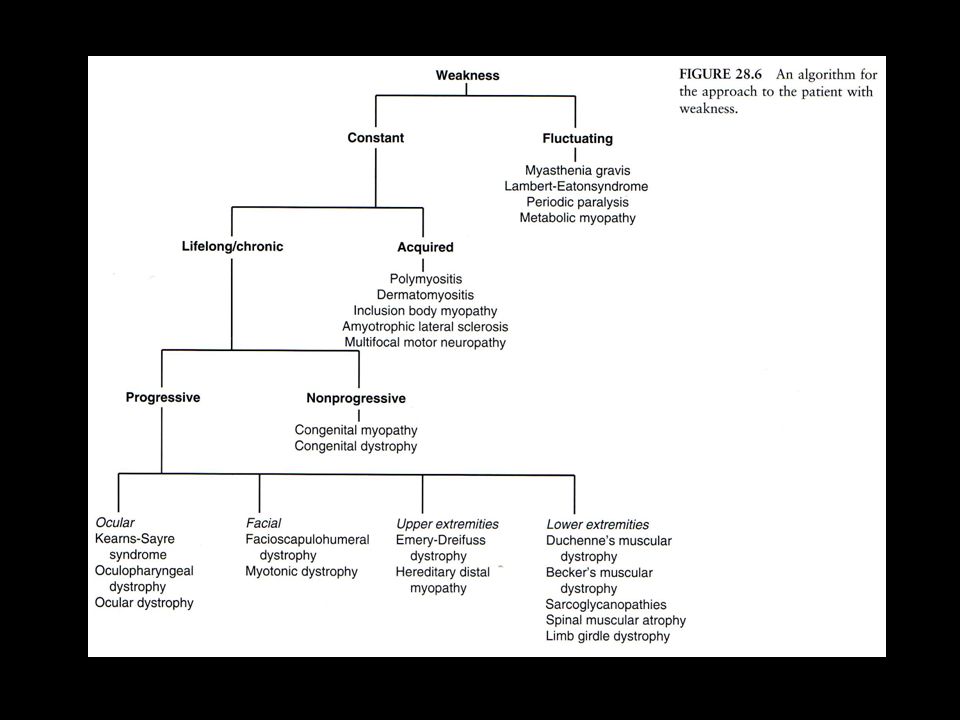
Evolution and duration
episodic periods of weakness with normal strength interictally (periodic paralysis, metabolic myopathies due to certain glycolytic pathway disorders). constant weakness
. constant weakness.")
23
Constant weakness acute or subacute progression
inflammatory myopathies (dermatomyositis and polymyositis); chronic slow progression over years most muscular dystrophies IBM nonprogressive weakness with little change over decades congenital myopathies
; chronic slow progression over years. most muscular dystrophies. IBM. nonprogressive weakness with little change over decades. congenital myopathies.")
24
Monophasic or relapsing
polymyositis can occasionally have an acute monophasic course with complete resolution of strength within weeks or months. Patients with periodic paralysis or metabolic myopathies can have recurrent attacks of weakness over many years, a patient with acute rhabdomyolysis due to cocaine may have a single episode.

25
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

26
Precipitating factors
illegal drug or prescription medication use that might produce a myopathy. weakness, pain, and/or myoglobinuria provoked by exercise a glycolytic pathway defect. Episodes of weakness with a fever carnitine palmityl transferase deficiency. Periodic paralysis is characteristically provoked by exercise or ingestion of a carbohydrate meal followed by a period of rest. Patients with paramyotonia congenita frequently report that cold exposure may precipitate their symptoms of muscle stiffness.

27
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

28
Are associated systemic symptoms or signs present? Cardiac disease
Arrhythmias Congestive Heart Failure Kearns-Sayre syndrome Andersen’s syndrome Duchenne Becker Polymyositis Emery-Dreifuss Muscular dystrophies Myotonic Limb-girdle 1B, 2C-2F, 2G Nemaline myopathy Emery-Dreifuss" Acid maltase deficiency Carnitine deficiency

29
Respiratory Insufficiency
Muscular Dystrophies Mitochondrial Myopathies Duchenne Becker Congenital Myopathies Emery-Dreifuss Nemaline Limb-girdle Centronuclear Myotonic Inflammatory Myopathies Congenital Metabolic Myopathies Polymyositis Acid maltase deficiency Carnitine deficiency

30
Hepatomegaly may be seen in myopathies associated with deficiencies in acid maltase, debranching enzyme, infectious disease. mitochondrial disorder.

31
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

32
Family history A thorough family history is clearly of great importance in making a correct diagnosis. Questions regarding family members’ use of canes or wheelchairs, skeletal deformities, or functional limitations are usually more informative than questions such as, ‘‘Does any member of your family have a muscle disease?’’

33
Diagnosis of Myopathy Based on Pattern of Inheritance
X-Linked Periodic paralysis Paramyotonia congenita Duchenne Thomsen disease Becker Emery-Dreifuss Central core myopathy" Autosomal Recessive Autosomal Dominant Facioscapulohumeral Metabolic myopathies Limb-girdle Oculopharyngeal muscular dystrophy Becker myotonia Maternal Transmission Myotonic dystrophy Mitochondrial myopathies

34
What is the distribution of weakness?
Which negative and/or positive symptoms and signs does the patient demonstrate? Family history History What is the temporal evolution? Are associated systemic symptoms or signs present? Are there precipitating factors that trigger episodic weakness or myotonia?

35
What is the distribution of weakness?
it is important to know which muscles to test and how to grade their power

36
Inspection Atrophy Pseudohypertrophy Fasciculation- MND or neuropathy
hypothyroid myopath selective atrophy of the quadriceps and forearm flexor muscles is highly suggestive of inclusion body myositis. Pseudohypertrophy Duchenne and Becker dystrophy Fasciculation- MND or neuropathy Distal myopathies may have profound atrophy of the anterior or posterior lower extremity compartments. Scapular wining FSHD. (LGMD 2G in 50% of pt) LGMD1B (laminopathy), LGMD2A (calpainopathy), LGMD2C–2F (sarcoglycanopathies). Hypertrophy LGMD2C–2F, LGMD2I, (LGMD 2G in 50% of pt) myotonia congenita amyloidosis, sarcoidosis, and Focal muscle enlargement can also be due to a neo- plastic or inflammatory process, ectopic ossification, tendon rupture, or partial denervation.
 LGMD1B (laminopathy), LGMD2A (calpainopathy), LGMD2C–2F (sarcoglycanopathies). Hypertrophy. LGMD2C–2F, LGMD2I, (LGMD 2G in 50% of pt) myotonia congenita. amyloidosis, sarcoidosis, and. Focal muscle enlargement can also be due to a neo- plastic or inflammatory process, ectopic ossification, tendon rupture, or partial denervation.")
38
Prominent Neck Extensor Weakness Proximal Limb-Girdle Weakness
PATTERN-RECOGNITION APPROACH TO MYOPATHIC DISORDERS Ptosis With or Without Ophthalmoplegia Distal Weakness Distal Arm/ProximalLeg Weakness Proximal Arm/Distal Leg Weakness

40
Pattern 1: Proximal Limb-Girdle Weakness
The most common pattern of muscle weakness in myopathies symmetrical weakness affecting predominantly the proximal muscles of the legs and arms The distal muscles are usually involved, but to a much lesser extent. Neck extensor and flexor muscles are also frequently affected. This pattern of weakness is seen in most hereditary and acquired myopathies and therefore is the least specific in ar- riving at a particular diagnosis

41
Pattern 2: Distal Weakness
This pattern of weakness predominantly involves the distal muscles of the upper or lower extremities (anterior or posterior compartment muscle groups) The involvement is usually, although not invariably, symmetrical. more commonly a feature of neuropathies
 The involvement is usually, although not invariably, symmetrical. more commonly a feature of neuropathies.")
42
Myopathies Characterized by Predominantly Distal Weakness
Distal Myopathies OPMD Late adult-onset distalmyopathy type 1 (Welander) Emery-Dreifuss Humeroperoneal Dystrophy*" Late adult-onset distal myopathy type 2 (Markesbery/Udd) Inflammatory Myopathies Inclusion Body Myositis Early adult-onset distal myopathy type 1 (Nonaka) Metabolic Myopathies Early adult-onset distal myopathy type 2 (Miyoshi) Debrancher deficiency Acid-maltase deficiency*" Early adult-onset distal myopathy type 3 (Laing) Congenital Myopathies Nemaline myopathy* Desmin myopathy Central core myopathy* Childhood-onset distal myopathy" Centronuclear myopathy Myotonic Dystrophy FSHD*" Scapuloperoneal Myopathy*" *" Scapuloperoneal pattern can occur
 Emery-Dreifuss Humeroperoneal Dystrophy* Late adult-onset distal myopathy type 2 (Markesbery/Udd) Inflammatory Myopathies. Inclusion Body Myositis. Early adult-onset distal myopathy type 1 (Nonaka) Metabolic Myopathies. Early adult-onset distal myopathy type 2 (Miyoshi) Debrancher deficiency. Acid-maltase deficiency* Early adult-onset distal myopathy type 3 (Laing) Congenital Myopathies. Nemaline myopathy* Desmin myopathy. Central core myopathy* Childhood-onset distal myopathy Centronuclear myopathy. Myotonic Dystrophy. FSHD* Scapuloperoneal Myopathy* * Scapuloperoneal pattern can occur.")
43
Pattern 3: Proximal Arm/Distal Leg Weakness
This pattern of weakness affects the periscapular muscles of the proximal arm and the anterior compartment muscles of the distal lower extremity (scapuloperoneal distribution) The scapular muscle weakness is usually characterized by scapular winging. Weakness can be very asymmetrical.
 The scapular muscle weakness is usually characterized by scapular winging. Weakness can be very asymmetrical.")
44
Myopathies with proximal arm/distal leg involvement (scapuloperoneal)
Scapuloperoneal dystrophy, Emery-Dreifuss dystrophy, LGMD1B, LGMD2A, LGMD2C–2F, Congenital myopathies, Nemaline myopathy Central core myopathy acid maltase deficiency. When this pattern is associated with facial weakness->(FSHD)
")
45
Pattern 4: Distal Arm/ProximalLeg Weakness
This pattern is associated with distal arm weakness involving the distal forearm muscles (wrist and finger flexors) and proximal leg weakness involving the knee extensors (quadriceps). The facial muscles are usually spared. Involvement of other muscles is extremely variable. The weakness is often asymmetrical between the two sides, which is uncommon in most myopathies. This pattern is essentially pathognomonic for inclusion body myositis (IBM). This pattern may also represent an uncommon presentation of myotonic dystropy; however, unlike IBM, muscle weakness is usually symmetrical
 and proximal leg weakness involving the knee extensors (quadriceps). The facial muscles are usually spared. Involvement of other muscles is extremely variable. The weakness is often asymmetrical between the two sides, which is uncommon in most myopathies. This pattern is essentially pathognomonic for inclusion body myositis (IBM). This pattern may also represent an uncommon presentation of myotonic dystropy; however, unlike IBM, muscle weakness is usually symmetrical.")
46
Pattern 5: Ptosis With or Without Ophthalmoplegia
usually, although not always, occurs without symptoms of diplopia. Facial weakness is not uncommon, extremity weakness is extremely variable, depending on the diagnosis.

47
The combination of ptosis, ophthalmoplegia (without diplopia), and dysphagia should suggest the diagnosis of oculopharyngeal dystrophy, especially if the onset is in middle age or later. Ptosis and ophthalmoplegia without prominent pharyngeal involvement is a hallmark of many of the mitochondrial myopathies. Ptosis and facial weakness without ophthalmoplegia is a common feature of myotonic dystrophy.

48
Myopathies With Ptosis or Ophthalmoplegia
Ptosis Without Ophthalmoplegia Ptosis With Ophthalmoplegia Myotonic dystrophy Congenital myopathies Centronuclear myopathy Nemaline myopathy Central core myopathy Desmin (myofibrillary) myopathy Oculopharyngeal muscular dystrophy Oculopharyngodistal myopathy Chronic progressive external ophthalmoplegia (mitochondrial myopathy) Neuromuscular junction disease (myasthenia gravis, Lambert-Eaton, botulism)
 myopathy. Oculopharyngeal muscular dystrophy. Oculopharyngodistal myopathy. Chronic progressive external ophthalmoplegia (mitochondrial myopathy) Neuromuscular junction disease (myasthenia gravis, Lambert-Eaton, botulism)")
49
Pattern 6: Prominent Neck Extensor Weakness
‘‘dropped head syndrome’’ Involvement of the neck flexors is variable. Isolated neck extension weakness represents a distinct muscle disorder called isolated neck extensor myopathy. Prominent neck extensor weakness: amyotrophic lateral sclerosis and myasthenia gravis.

50
Myopathies With Prominent Neck Extensor Weakness
Isolated neck extensor myopathy" Polymyositis " Dermatomyositis " Inclusion body myositis " Carnitine deficiency " Facioscapulohumeral dystrophy " Myotonic dystrophy " Congenital myopathy " Hyperparathyroidism

51
History Clinical evaluation Examination Investigations

52
Lab ( CK) Investigations Muscle Biopsy NCS/EMG Genetics
 Investigations Muscle Biopsy NCS/EMG Genetics")
53
Creatine Kinase The CK is elevated in the majority of myopathies but may be normal in slowly progressive myopathies. Duchenne dystrophy, the CK level is invariably at least 10 times (and often up to 100 times) normal. CK may also be markedly elevated: LGMD1C (caveolinopathy), LGMD2A (calpainopathy), and LGMD2B (dysferlinopathy), The CK level may not be elevated in corticosteroid administration, collagen diseases, alcoholism hyperthyroidism profound muscle wasting
 normal. CK may also be markedly elevated: LGMD1C (caveolinopathy), LGMD2A (calpainopathy), and. LGMD2B (dysferlinopathy), The CK level may not be elevated in. corticosteroid administration, collagen diseases, alcoholism. hyperthyroidism. profound muscle wasting.")
54
Differential Diagnosis of Creatine Kinase Elevation
Myopathies Medications Muscular dystrophies Hypothyroidism/ Hypoparathyroidism Congenital myopathies SurgeryTrauma (electromyography studies, intramuscular or subcutaneous injections) Metabolic myopathies Inflammatory myopathies Drug/toxin-induced Strenuous Exercise Carrier state (dystrophinopathies) Channelopathies Increased Muscle Mass Motor Neuron Diseases Race ALS Sex SMA ‘‘Idiopathic HyperCKemia’’ Postpolio syndrome Neuropathies GBS CIDP Viral Illness Race = ck often is above the normal range in some African American individuals and in patients with enlarged muscles.
 Metabolic myopathies. Inflammatory myopathies. Drug/toxin-induced. Strenuous Exercise. Carrier state (dystrophinopathies) Channelopathies. Increased Muscle Mass. Motor Neuron Diseases. Race. ALS. Sex. SMA. ‘‘Idiopathic HyperCKemia’’ Postpolio syndrome. Neuropathies. GBS. CIDP. Viral Illness. Race = ck often is above the normal range in some African American individuals and in patients with enlarged muscles.")
55
Electrophysiological Studies
Confirm localization can be a guide as to which muscle to biopsy. r/o neuropathy, NMG disease, MND. NCS are typically normal in patients with myopathy. Needle EMG examination: motor units Brief duration, small-amplitude Early recruitment Needle EMG can also provide a clue as to which muscles have had recent or ongoing muscle in- jury and can be a guide as to which muscle to biopsy. It is important to realize, however, that the EMG can be normal in a patient with myopathy, and the results of electrodiagnostic studies need to be evaluated in the context of the patient’s history, neurological ex- amination, and other laboratory studies.

56
Muscle biopsy Selection of the appropriate muscle to biopsy is critical. Muscles that are severely weak (MRC grade 3 or less) should not be biopsied, since the results are likely to show only evidence of end stage muscle disease muscles that have recently been studied by needle EMG should be avoided because of the possibility of artifacts created by needle insertion.
 should not be biopsied, since the results are likely to show only evidence of end stage muscle disease. muscles that have recently been studied by needle EMG should be avoided because of the possibility of artifacts created by needle insertion.")
57
Biopsies should generally be taken from muscles that demonstrate MRC grade 4 strength.
The muscle of choice: biceps. vastus lateralis. The gastrocnemius should be avoided, since its tendon insertion extends throughout the muscle and inadvertent sampling of a myotendinous junction may cause difficulty with interpretation.

58
Typical myopathic abnormalities include:
central nuclei, both small and large hypertrophic round fibers, split fibers, and degenerating and regenerating fibers. Chronic myopathies frequently show evidence of increased connective tissue and fat.

59
Inflammatory myopathies
characterized by: the presence of mononuclear inflammatory cells in the endomysial and perimysial connective tissue between fibers and occasionally around blood vessels ( perivascular)
")
60
Dermatomyositis …….In addition,
perifascicular atrophy, characterized by atrophy of fibers located on the periphery of a muscle fascicle, is a common finding. Inflammatory cell invasion of nonnecrotic fibers is not prominent. deposition of the C5b-9 or membrane attack complex on or around small blood vessels

61
polymyositis variability in fiber size,
scattered necrotic and regenerating fibers, endomysial inflammation with invasion of non-necrotic muscle fibers

62
IBM endomysial inflammation with invasion of non-necrotic muscle fibers small groups of atrophic fibers, eosinophilic cytoplasmic inclusions, and muscle fibers with one or more rimmed vacuoles lined with granular material Amyloid deposition is evident on Congo red staining. TDP-43 positive Ubiquitin positive Electron microscopy demonstrates 15-nm to 21-nm cytoplasmic and intranuclear tubulofilaments

64
Mitochondrial disorder
The typical muscle histopathology is “ragged red’’ fibers subsarcolemmal accumulation of abnormal mitochondria stains red. best seen with modified–Gomori trichrome stain

65
Type 1 fibers (slow-twitch, fatigue-resistant, oxidative metabolism) stain lightly at alkaline and darkly at acidic pH levels. Type 2 fibers (fast- twitch, fatigue-prone, glycolytic metabolism) stain darkly at alkaline and lightly at acidic pH levels. Normally, a random distribution of the two fiber types occurs, and generally twice as many type 2 as type 1 fibers are identified.
 stain darkly at alkaline and lightly at acidic pH levels. Normally, a random distribution of the two fiber types occurs, and generally twice as many type 2 as type 1 fibers are identified.")
67
W Increased variability in muscle fiber size degeneration, regeneration, isolated ‘‘opaque’’ hypertrophic fibers, and significant replacement of muscle by fat and connective tissue. Central nuclei are present in 2% to 4% of the fibers. Type 1 fiber predominance is seen in most patients. Inflammatory cell infiltrates in the perimysium, endomysium, and perivascular spaces and consist mostly of mononuclear cells, especially macrophages. endomysial and perimysial fibrosis with disease progression

68
A, C, D, Muscle biopsy of Duchenne muscular dystrophy and (B) normal control. By light microscopy, the biopsy demonstrates marked variability in fiber size,degenerating and regenerating fibers, ‘‘opaque’’ hypertrophic fibers, and replacement of muscle by fat and connective tissue (most findings shown in A and C). Note the lack of these findings in a normal muscle biopsy (B). In C, arrows show small regenerating muscle fibers and a necrotic fiber invaded by mononuclear cells (open arrow). In D, arrows indicatethe dark-staining ‘‘opaque’’ or hypercontracted large fibers. A, B, D, Hematoxylin and eosin; C, modified trichrome.
. Note the lack of these findings in a normal muscle biopsy (B). In C, arrows show small regenerating muscle fibers and a necrotic fiber invaded by mononuclear cells (open arrow). In D, arrows indicatethe dark-staining ‘‘opaque’’ or hypercontracted large fibers. A, B, D, Hematoxylin and eosin; C, modified trichrome..")
69
Immunostaining of frozen sections of skeletal muscle biopsies
Immunostaining of frozen sections of skeletal muscle biopsies. D through N show antidystrophin antibodies using indirect immunofluorescence. A, B, and C display hematoxylin andeosin–stained sections corresponding to D, E, and F respectively. There is complete absence of immunofluorescence at the sarcolemma in a muscle section from a patient with Duchenne muscular dystrophy (DMD) (C and F), compared with the homogeneous staining of the plasma membrane in normal muscle (A and D) and in muscle biopsies from patients with Emery-Dreifuss muscular dystrophy (G), Fukuyama type of congenital muscular dystrophy (H), limb-girdle muscular dystrophy (I), facioscapulohumeral muscular dystrophy (J), myotonic dystrophy (K), and Kugelberg-Welander type of spinal muscular atrophy (L). In a frozen section of muscle from a patient with BMD (B), partial staining of the sarcolemma is observed (E). Note the mosaic pattern of immunostaining in muscle biopsies from a symptomatic (M) and an asymptomatic (N) DMD carrier.
 (C and F), compared with the homogeneous staining of the plasma membrane in normal muscle (A and D) and in muscle biopsies from patients with Emery-Dreifuss muscular dystrophy (G), Fukuyama type of congenital muscular dystrophy (H), limb-girdle muscular dystrophy (I), facioscapulohumeral muscular dystrophy (J), myotonic dystrophy (K), and Kugelberg-Welander type of spinal muscular atrophy (L). In a frozen section of muscle from a patient with BMD (B), partial staining of the sarcolemma is observed (E). Note the mosaic pattern of immunostaining in muscle biopsies from a symptomatic (M) and an asymptomatic (N) DMD carrier.")
70
Nemaline myopathy Type I fiber predominance and atrophy
Marked disproportion in fiber size, with uniformly small type I fibers and normal to large type II fibers Sarcoplasmic rods (nemaline bodies) Short, granular-appearing Subsarcolemmal and perinuclear localization in type I fibers (especially large rods) Composed of α-actinin, actin, and other Z diskproteins, and appear to arise from, are attached to,and thicken the Z disk (small rods) Intranuclear rods
 Short, granular-appearing. Subsarcolemmal and perinuclear localization in type I fibers (especially large rods) Composed of α-actinin, actin, and other Z diskproteins, and appear to arise from, are attached to,and thicken the Z disk (small rods) Intranuclear rods.")
71
Myotubular (centronuclear) Myopathy
Is called “centronuclear” because of predominance of small type I fibers with central nucleib. Type I fiber predominance (often small, atrophied) Central pallor noted on ATPase stainingd. Radial distribution of sarcoplasmic strands apparent on NADH reactione. Interstitial connective tissue: normal or mildly increased
 Central pallor noted on ATPase stainingd. Radial distribution of sarcoplasmic strands apparent on NADH reactione. Interstitial connective tissue: normal or mildly increased.")
72
Central Core Disease Type I fiber predominance Central cores
Single, well-circumscribed regions Selectively involve type I fibers Deficient in mitochondria and sarcoplasmic reticulum as well as oxidative enzymes and phosphorylase activity (detected best on sections with oxidative enzyme immunohistochemistry ,as poorly stained central demarcated areas) Extend entire length of muscle fiber Increased endomysial connective tissue. Often, increased number of internal nuclei
 Extend entire length of muscle fiber. Increased endomysial connective tissue. Often, increased number of internal nuclei.")
73
Myofibrillar myopathy
Myofibrillar myopathy. A and B, Trichrome-stained sections show aggregates of dense granular and filamentous amor- phous material and vacuolar change. Immunohistochemistry performed in sections corresponding to that in B demonstrates accumulation of desmin (C) and αB-crystallin (D).
 and αB-crystallin (D).")
74
Molecular Genetic Studies
Disorders With Commercially Available Molecular Genetic Studies Performed With Peripheral Blood Samples Duchenne and Becker muscular dystrophies ” FSHD MD (types 1 and 2) " OPMD LGMD1B, 2A, 2C–2F, and 2I " Congenital muscular dystrophy (FKRP, FCMD, MEB, and POMT1 mutations) " Nonaka myopathy/inclusion body myopathy type 2 " Nemaline myopathy (ACTA1 mutations) " Myotubular myopathy (MTM1 mutations) " MERRF MELAS
 OPMD. LGMD1B, 2A, 2C–2F, and 2I Congenital muscular dystrophy (FKRP, FCMD, MEB, and POMT1 mutations) Nonaka myopathy/inclusion body myopathy type 2 Nemaline myopathy (ACTA1 mutations) Myotubular myopathy (MTM1 mutations) MERRF. MELAS.")
76
Case 1 A 51-year-old man without significant past medical Slowly progressive muscle weakness for the past 7 years. His symptoms initially began with difficulty walking down stairs because his left knee would ‘‘give out.’’ He currently has difficulty arising from a chair and grasping objects with his right hand.

78
Current examination reveals intact cranial nerves, sensation, and muscle stretch reflexes.
Motor examination in the right upper extremity shows MRC grade 5 shoulder abduction, grade 5 elbow flexion/extension, grade 4 wrist flexion, grade 5 wrist extension, and grade 3 finger flexion. Strength in the left upper extremity is normal except for grade 4+ finger flexion. In the left lower extremity, the patient exhibits grade 4+ hip flexion, grade 3+ knee extension, and grade 4+ ankle dorsiflexion. In the right lower extremity, strength is normal except for grade 4+ knee extension.

79
The chronic onset, asymmetrical distribution of weakness, and selective involvement of wrist/finger flexion and knee extension CK= 500 EMG= myopathy Muscle biopsy

80
IBM

81
Case 2 Describe and give DDx.
Muscle involved: serratus anterior or long thoracic nerve Weakness of trapezius causes winging of scapula when the arm is abducted. Also seen in LGMD

82
FSHD normal deltoids biceps, triceps commonly weak, forearm muscles are less involved resulting in a popeye-like appearance wrist flexors stronger than wrist extensors hip flexors and quadriceps are weaker, plantar flexors are preserved, ankle dorsiflexion is weak and foot drop may be the presenting symptom Scapular winging may be very asymmetric (misdiagnosed as long thoracic nerve of Bell palsy) The genetic abnormality is a deletion in a 3.3-kb repeating sequence termed D4Z4. Chromosome 4
 The genetic abnormality is a deletion in a 3.3-kb repeating sequence termed D4Z4. Chromosome 4.")
83
Thanks!

Similar presentations












 Wenya Hou Xue Jing Yitang Wang Jiezhong Zhang.>")
 is a group of inherited muscle diseases, in which muscle fibers are unusually susceptible to damage. Muscles, primarily voluntary.>")
 Unlike: neuronopathies: secondary.>")










