Download presentation
Presentation is loading. Please wait.

1
Mechanisms of Leukemogenesis in Patients with SCN Daniel C. Link
Clonal dominance Role of alterations in the bone marrow microenvironment

2
Severe Congenital Neutropenia (Kostmann’s Syndrome)
Clinical manifestations: Chronic severe neutropenia present at birth Accumulation of granulocytic precursors in the bone marrow Recurrent infections Treatment with G-CSF Reduces infections and improves survival Marked propensity to develop acute myeloid leukemia or myelodysplasia

3
Stem Cell CFU-GM What are the molecular mechanisms for the isolated block in granulopoiesis Myeloblast Promyelocyte Block in granulocytic differentiation Myelocyte What is the molecular basis for the marked susceptibility to AML Metamyelocyte Band Neutrophil Segmented Neutrophil

4
Genetics of SCN

5
ELANE Mutations All mutations are heterozygous
Act in a cell intrinsic fashion to inhibit granulopoiesis

6
Molecular Pathogenesis of SCN associated with ELANE Mutations
Working hypothesis: ELANE mutations lead to the production of misfolded neutrophil elastase, induction of the unfolded protein response, and the subsequent apoptosis of granulocytic precursors resulting in neutropenia.

7
SCN and MDS/AML Cumulative risk of MDS/AML in SCN: 21% after treatment with G-CSF for 10 years Cumulative risk of leukemia (all types) up to age 40: 0.15%
 up to age 40: 0.15%")
8
Risk of AML/MDS in Bone Marrow Failure Syndromes

9
G-CSFR Mutations in SCN
G-CSF receptor Member of cytokine receptor superfamily Only known receptor for G-CSF G-CSF receptor mutations in SCN Acquired heterozygous mutations Strongly associated with the development of AML Box 1 Box 2 C -Y G-CSFR d715

10
Questions Do the G-CSFR mutations contribute to leukemic transformation? And if so, How do the G-CSFR mutations gain clonal dominance? What are the molecular mechanisms

11
d715 “Knock-in” Mice WT G-CSFR gene Targeting vector
Stop codon d715 G-CSFR allele d715 mice have normal basal granulopoiesis

12
d715 Tumor Watch 100 200 300 400 25 50 75 WT/WT WT/d715 WT/WT + G-CSF WT/d715 + G-CSF Time (days) Percentage survival The d715 G-CSFR is not sufficient to induce in mice even with chronic G-CSF stimulation
 Percentage survival. The d715 G-CSFR is not sufficient to induce in mice even with chronic G-CSF stimulation.")
13
Oncogene Cooperativity
Growth Factor Mutations Transcription Factor + FLT3 ITD PML-RARα + d715 G-CSFR PML-RARα + Leukemia?

14
D715 G-CSFR Tumor Watch Truncations mutations of the G-CSFR contribute to leukemic transformation in SCN.

15
G-CSFR mutations may be an early event during leukemogenesis
(age-years) SCN SCN G-CSFR SCN G-CSFR Runx1 AML G-CSFR Runx1 -7, 5q-
 SCN. SCN. G-CSFR. SCN. G-CSFR. Runx1. AML. G-CSFR. Runx1. -7, 5q-")
16
Clonal Dominance G-CSFR mutations Clinical Leukemia
Likely has to occur in a long-lived self-renewing cell (eg, stem cell)
")
17
Competitive Repopulation Assay
Wild type 1:1 Ratio Harvest Bone Marrow Wild type d715 d715 1,000 cGy Bone Marrow Chimera Syngeneic Recipient wild type (Ly5.1)
")
18
Competitive Repopulation Assay
Wild type d715 3-6 Months No Competitive Advantage Competitive Advantage

19
Donor Chimerism Analysis
B Lymphocytes Neutrophils B220 Gr-1 61.8% 51.0% Ly5.2 (d715) Ly5.2 (d715)
 Ly5.2 (d715)")
20
6 months after transplantation—1:1 ratio
d715 Chimeras 6 months after transplantation—1:1 ratio Red blood cell Platelet Common Myeloid Progenitor Neutrophil Monocyte Common Lymphoid Progenitor CFU-GM BFU-E CFU-Meg B cell HSC T cell 63.5% 46.6% 50.0% 45.7%

21
d715 Chimeras G-CSF (10ug/kg/d x 21 days)
Red blood cell Platelet Common Myeloid Progenitor Neutrophil Monocyte Common Lymphoid Progenitor CFU-GM BFU-E CFU-Meg B cell HSC T cell BM 63.3% 89.1% 75.8% 98.6% 61.1% 68.4% 49.7% 60.5% 52.6% 97.6%

22
Long-term d715 G-CSFR chimerism following G-CSF treatment for 21 days
69.2 76.6 47.3 56.9

23
d715 Chimeras G-CSF (10ug/kg/d x 21 days)
53.3% 97.8% Red blood cell Platelet Common Myeloid Progenitor Neutrophil Monocyte Common Lymphoid Progenitor CFU-GM BFU-E CFU-Meg B cell HSC T cell BM 63.3% 89.1% 75.8% 98.6% 61.1% 68.4% 49.7% 60.5% 52.6% 97.6%

24
Conclusion The d715-G-CSFR confers a clonal advantage at the hematopoietic stem cell level in a G-CSF dependent fashion

25
RNA Expression Profiling
WT d715 G-CSF Saline G-CSF Saline Harvest bone marrow at 3 hours Sort Kit+ Sca+ Lineage- (KSL) cells RNA expression profiling
 cells. RNA expression profiling.")
27
In mutant GR KSL cells, STAT3 activation by G-CSF is attenuated while STAT5 activation is enhanced
Stat3 phosphorylation Stat5 phosphorylation

28
G-CSFR mutations Clonal Dominance Acts at the HSC level Dependent on exogenous G-CSF Mediated by exaggerated STAT5 activation What are the STAT5 target genes that mediate clonal dominance Would inhibitors of STAT5 (or their target genes) be effective therapeutic agents in AML.
 be effective therapeutic agents in AML.")
29
Stem Cell Niches Osteoblast Niche Vascular Niche

30
Chronic disruption of the stem cell niche in the bone marrow may contribute to the high rate of leukemic transformation in bone marrow failure syndromes Normal BMFS (e.g., SCN) G-CSF high G-CSF low
 G-CSF high. G-CSF low.")
31
G-CSF ROS induction is rapid in vitro
(within minutes) Wild-type d715 G-CSFR Prolonged G-CSF (≥ 5 days) is associated with marked changes in bone marrow stromal cells Single dose G-CSF 7 days of G-CSF No G-CSF Harvest Bone Marrow Flow Cytometry ROS in KSL cells H2AX phosphorylation in KSL cells
 Wild-type. d715 G-CSFR. Prolonged G-CSF (≥ 5 days) is associated with marked changes in bone marrow stromal cells. Single dose. G-CSF. 7 days of. G-CSF. No G-CSF. Harvest Bone Marrow. Flow Cytometry. ROS in KSL cells. H2AX phosphorylation in KSL cells.")
32
ROS Induction is increased in d715 KSL cells after 7 days of GCSF Rx

33
H2Ax Phosphorylation Enhanced in d715 KSL cells after 7 days of GCSF Rx
We next measured H2AX phosphorylation. H2AX phosphorylation is an early and sensitive marker of DNA damage. <click> In wild type mice, no increase in H2AX-P was observed after short of prolonged G-CSF administration. <cllick> In d715 G-CSFR mice, the basal level of H2AX-P was low. Again, short term G-CSF treatment did not significantly induced H2AX-P, while 7 days of G-CSF resulted in a significant increase.

34
NAC attenuates G-CSF induced H2AX phosphorylation
G-CSF (7 days) alone Measurement ROS H2AX-P G-CSF (7 days) + N-acetyl cysteine (NAC) WT or d715 G-CSFR mice
 alone. Measurement. ROS. H2AX-P. G-CSF (7 days) + N-acetyl cysteine (NAC) WT or d715 G-CSFR mice.")
35
Hypothesis: Changes in the BM microenvironment induced by G-CSF contribute to DNA damage
. G-CSF treatment in mice Decreases osteoblasts Decreases SDF1 expression These effects are delayed, first becoming apparent on day of G-CSF

36
G-CSF suppresses mature osteoblasts
Untreated G-CSF As I mentioned, osteoblast lineage cells are a key cellular component of the stem cell niche. We and others showed that G-CSF treatment is associated with marked suppression of osteoblasts in the bone marrow. Shown here is representative photomicrograph of the femur of a transgenic mouse that expresses EGFP in osteoblasts. The green cells line the endosteal and trabecular bone surfaces. <click> Treatment of mice with G-CSF for 7 days results in a marked loss of osteoblasts in the bone marrow.

37
Signaling through the d715 G-CSFR results in marked osteoblast and CXCL12 (SDF1) suppression
 suppression")
38
Question: Does disruption of stromal/HSPC interactions sensitize cells to G-CSF induced oxidative DNA damage Normal AMD3100 G-CSF low Specific CXCR4 antagonist Disrupts HSPC/stromal interactions Results in HSPC mobilization

39
Question: Does disruption of stromal/HSPC interactions sensitize cells to G-CSF induced oxidative DNA damage G-CSF (1 dose) alone Measure H2AX phosphorylation G-CSF (1 dose) + AMD3100 WT or d715 G-CSFR mice
 alone. Measure. H2AX phosphorylation. G-CSF (1 dose) + AMD3100. WT or d715 G-CSFR mice.")
40
SCN Normal G-CSF low G-CSF high
. Lowering G-CSF levels (by treating the underlying neutropenia) may reduce the risk of AML Biomarkers of bone metabolism might predict risk of AML Treatment with G-CSF, by disrupting the stem cell niche, may sensitize leukemic cells to chemotherapy
 may reduce the risk of AML. Biomarkers of bone metabolism might predict risk of AML. Treatment with G-CSF, by disrupting the stem cell niche, may sensitize leukemic cells to chemotherapy.")
41
The article I am going to present is titled, Genetic variegation of clonal architecture and prropogating cells in leukemia, published this month in nature. This is a relative short article and so I am going to intially talk about the background to put this article in perspective Nature, Jan 20, 2011

42
5-year survival rate of 80% Structural abnormalities:
Pre-B ALL is the most common pediatric cancer – 30% of all cancers in children 5-year survival rate of 80% Structural abnormalities: t(12;21) ETV6/RUNX1 : 20-25% t(1;19) E2A/PBX1 translocation: 5 % t(4;11) MLL/AF4 rearrangement : 5% t(9;22) BCR/ABL translocation (Philadelphia chromosome): 3-4% t(8;14) MYC/IGH translocation : 1% In addition to hyperdiploidy and hypodiploidy there are several specific chromosoomal translocation descrbed in childhood ALL.
 ETV6/RUNX1 : 20-25% t(1;19) E2A/PBX1 translocation: 5 % t(4;11) MLL/AF4 rearrangement : 5% t(9;22) BCR/ABL translocation (Philadelphia chromosome): 3-4% t(8;14) MYC/IGH translocation : 1% In addition to hyperdiploidy and hypodiploidy there are several specific chromosoomal translocation descrbed in childhood ALL.")
43
Subset of childhood pre-B ALL with ETV6-RUNX1 fusion
ETV6-RUNX1 fusion as we have discussed is predominantly a prenatal event and a presumed initiating event. This si asssociated with modest number of recurrent genomic CAN. Del of ETV6 is the most common present in present in 97% of the cases. Other common events are Zelent, Oncogene, 2004 Associated with modest number of recurrent genomic CNA (3-6). Del ETV6, del CDKN2A, del PAX5, del 6q, gain Xq
. Del ETV6, del CDKN2A, del PAX5, del 6q, gain Xq.")
44
Figure 1A Here is an example of an apparent linear architecture with 3 clones. Describe the events….

45
Figure 1B Moderately complex architecture with 5 subclones… Loss of ETV6… Loss of both CDK and then a loss of ETV6. Here there is a gain of RUNX and then a loss of ETV6.

46
Figure 1C

47
Author comments Common or highly recurrent CNA are not acquired in any particular order. Sub-clones with highest number of CNA were not necessarily numerically dominant. CNA involving the same gene could be simultaneously present in distinct sub-clones and must therefore arise more than once, independently.

48
Supplementary Figure 3

49
Supplementary Figure 3

50
Figure 2A

51
Figure 2b

52
Supplementary Figure 2

53
Author Comments Clonal architecture at relapse is different from that of diagnosis in most patients. Relapse seem to derive from either major or minor clones at diagnosis but with a suggestion that more than one sub-clone might contribute to relapse. The dominant sub-clone in relapse itself continues to genetically diversify.

54
Xenotransplantation Assay
Secondary transplant 2x equivalent ALL cells 2x Unfractionated or immunophenotypically Flow sorted ALL primary cells NOD/SCID IL2Rγnull NOD/SCID IL2Rγnull 250 cGy 250 cGy

55
Figure 3 Patient 3

56
Here is the summary table of different serial transsplants done with patient no,3 sample. The diagnostic samples clonesa re in the top row. One of the clones is mostly not detecctable, some clones are lost and the others are preserved.

57
Figure 4a-c Patient 7 Here 2 of the clones decreased following serial transsplantation. And one of the smaller clones explanded. And the primary clone not detectable anymore.

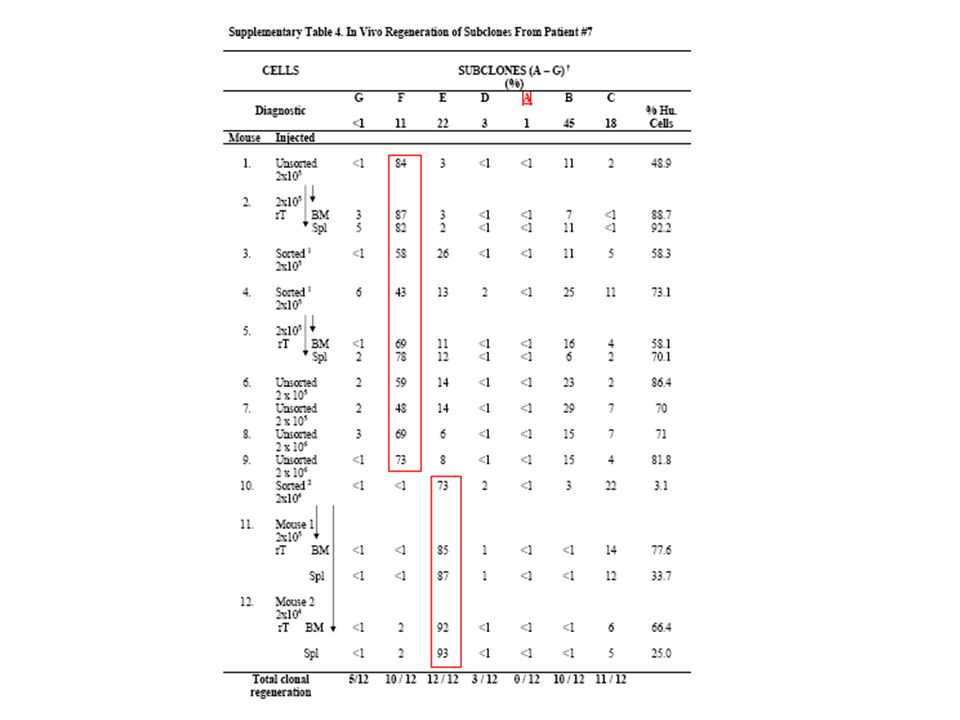
59
Figure 4d Patient 3 They show some SNP array data for CAN here. There were shred CAN between the samples and some distinct ones.

60
Author Summary Distinctive genotypes are associated with variable capacity for leukemia propagation. The relevance of the xenotransplantation to study clonal expansion is questionable. Studies of clonal evolution in patients with ALL (e.g., at diagnosis and at relapse) are more relevant.
 are more relevant.")
61
Comments Clonal diversity is underestimated in this study (only few CNAs were measured. Not particularly sensitive assay with 1% detectable threshold. To study the full complexity of subclonal architecture will require whole genome genome sequencing at single cell level ( or colonies from leukemic cells). Implications for targeted therapy in cancer…
. Implications for targeted therapy in cancer…")
Similar presentations

 (q22;q21) developing inv(16)(p13q22) secondary AML.>")
 gene, one of the most common gene mutations (25%-30%) in AML NPM1 mut co-occurs.>")




. A closer look at the E2A gene... Other names: TCF3, ITF1, and Factors E12/E47 Located on chromosome 19 Encodes.>")
 by Lindsey Wilfley.>")

>")


 to those.>")
>")





