Download presentation
Presentation is loading. Please wait.

1
LEUKAEMIA DIAGNOSIS

2
Leukaemia is a disease resulting from the neoplastic proliferation of haemopoietic
or lymphoid cells. Leukaemias are broadly divided into: Acute leukaemias, which, if untreated, lead to death in weeks or months. chronic leukaemias, which, if untreated, lead to death in months or years.

3
They are further divided into lymphoid, myeloid and biphenotypic leukaemias, the latter showing both lymphoid and myeloid differentiation. Acute leukaemias are characterized by a defect in maturation, leading to an imbalance between proliferation and maturation; since cells of the leukaemic clone continue to proliferate without maturing to end cells.

4
Diagnosis of leukaemia.
The diagnosis of leukaemia and categorization required the following parameters. 1- Morphology. 2-Cytochimestry 3-Immunophenotyping. 4-Cytogenetic. 5- Molecular study.

5
Acute Lymphoblastic Leukaemia

6
Background Acute lymphoblastic leukemia (ALL) is the most common malignancy diagnosed in children, representing nearly one third of all pediatric cancers. The annual incidence rate for acute lymphoblastic leukemia is 30.9 cases per million population. The peak incidence occurs in children aged 2-5 years.
 is the most common malignancy diagnosed in children, representing nearly one third of all pediatric cancers. The annual incidence rate for acute lymphoblastic leukemia is 30.9 cases per million population. The peak incidence occurs in children aged 2-5 years.")
7
Pathophysiology In acute lymphoblastic leukemia, a lymphoid
progenitor cell becomes genetically altered and subsequently undergoes dysregulated proliferation, survival, and clonal expansion. In most cases, the pathophysiology of transformed lymphoid cells reflects the altered expression of genes whose products contribute to the normal development of B cells and T cells

8
Clinical feature. Children with acute lymphoblastic leukemia (ALL) generally Present with signs and symptoms that reflect bone marrow infiltration and extramedullary disease. Because leukemic blasts replace the bone marrow, patients Present with signs of bone marrow failure, including anemia, thrombocytopenia, and neutropenia. Clinical manifestations include fatigue and pallor, petechiae and bleeding, and fever. In addition, leukemic spread may manifest as lymphadenopathy And hepatosplenomegaly. Other signs and symptoms of leukemia including weight loss, bone pain, and dyspnea.

9
The classification of ALL
FAB classification. L1 ALL L2 ALL L3 ALL Cell size Mainly small Large, heterogeneous Large, homogeneous Nuclear chromatin Fairly homogeneous Heterogeneous Finely stippled, Nuclear shape Mainly regular Irregular; clefting Regular Nucleolus Not visible Usually visible Usually prominent Amount of cytoplasm Scanty Variable abundant Moderately abundant Cytoplasmic basophilia Slight to moderate Variable Strong Cytoplasmic vacuolation Variable Variable Often prominent

14
Clinical correlates of FAB categories of ALL
Many cases of L3 ALL represent a distinct entity that requires specific management. However, the categorization of a case as L1 or L2 ALL is of little importance. The FAB L1 category includes more childhood cases with a relatively good prognosis. The incidence of ALL L1 falls with increasing age whereas the incidence of ALL L2 does not vary much with age. ALL L2 has generally been found to have a worse prognosis, although the difference is not major.

15
WHO proposed classification of acute lymphoblastic leukemia
The recent WHO International panel on ALL recommends that the FAB classification be abandoned, since the morphological classification has no clinical or prognostic relevance. 1- Acute lymphoblastic leukemia/lymphoma Synonyms: Former Fab L1/L2 i. Precursor B acute lymphoblastic leukemia/lymphoma. Cytogenetic subtypes: t(12;21)(p12,q22) TEL/AML-1 t(1;19)(q23;p13) PBX/E2A t(9;22)(q34;q11) ABL/BCR T(V,11)(V;q23) V/MLL ii. Precursor T acute lymphoblastic leukemia/lymphoma 2- Burkett's leukemia/lymphoma Synonyms: Former FAB L3 3- Biphenotypic acute leukemia
(p12,q22) TEL/AML-1. t(1;19)(q23;p13) PBX/E2A. t(9;22)(q34;q11) ABL/BCR. T(V,11)(V;q23) V/MLL. ii. Precursor T acute lymphoblastic leukemia/lymphoma. 2- Burkett s leukemia/lymphoma Synonyms: Former FAB L3. 3- Biphenotypic acute leukemia.")
16
Immunophenotyping

17
Characterization of the Immunophenotyping is referred to as Immunophenotyping and is achieved by means of labeled antibodies that recognize specific epitopes of cellular antigens. In general, the most useful antibodies are monoclonal antibodies (McAb) produced by hybridoma technology but, for some antigens, polyclonal antibodies (PcAb) (antisera) are better. The technique employed for Immunophenotyping may be immunocytochemistry or, much more often, flow cytometry. Immunophenotyping is essential for the diagnosis of B- or T-lineage acute lymphoblastic leukaemia (ALL).
 produced by hybridoma technology but, for some antigens, polyclonal antibodies (PcAb) (antisera) are better. The technique employed for Immunophenotyping may be immunocytochemistry or, much more often, flow cytometry. Immunophenotyping is essential for the diagnosis of B- or T-lineage acute lymphoblastic leukaemia (ALL).")
18
First panel Second panel B lymphoid CD19, CD22, CD79a, CD10
T lymphoid CD3, CD2, CD7 Second panel If B lineage cm, k, l, CD20, CD24 If T lineage CD1a, SmCD3, CD4, CD5, CD8, anti-TCR ab, anti-TCR gd

20
Cytogenetic study. With techniques now available, 70–90% of cases of ALL have a demonstrable cytogenetic abnormality. In ALL, chromosomal abnormalities correlate with other clinical and hematological factors of prognostic importance but they also have a considerable independent prognostic significance.

21
B-lineage ALL L1 high/ hyperdiploidy. L1 or L2/t(9;22)/BCR-ABL fusion
L1 or L2/t(4;11)(q21;q23) L1 or L2/t(12;21)(p12;q22)/early precursor or common ALL L1 or L2/t(1;19)(q23;p13)/pre-B ALL
(q21;q23) L1 or L2/t(12;21)(p12;q22)/early precursor or common ALL. L1 or L2/t(1;19)(q23;p13)/pre-B ALL.")
22
T-lineage ALL. L1 or L2/t(10;14)(q24;q11) Burkett's-lineage
L3/t(8;14)(q24;q32) or t(8;22)(q24;q11) or t(2;8)(p12;q24).
(q24;q32) or t(8;22)(q24;q11) or. t(2;8)(p12;q24).")
23
Acute Myeloblastic Leukaemia

24
Distinguishing between AML and ALL
Correct assignment of patients to the categorize AML and ALL is very important for prognosis and choice of therapy. The FAB group recommended the use of MPO,SBB and non-specific esterase (NSE) stains. If Cytochemical reactions for myeloid cells are negative, presumptive diagnosis of ALL must be confirmed Immunophenotyping.
 stains. If Cytochemical reactions for myeloid cells are. negative, presumptive diagnosis of ALL must. be confirmed Immunophenotyping.")
25
Background. AML is the most common acute leukaemia affecting adults, and its incidence increases with age. Although AML is a relatively rare disease, accounting for approximately 1.2% of cancer deaths in the United States, its incidence is expected to increase as the population ages.

26
The malignant cell in AML is the myeloblast.
Pathophysiology. The malignant cell in AML is the myeloblast. In normal haematopoiesis, the myeloblast is an immature precursor of myeloid white blood cells; a normal myeloblast will gradually mature into a mature white blood cell. However, in AML, a single myeloblast accumulates genetic changes which "freeze" the cell in its immature state and prevent differentiation Such a mutation alone does not cause leukemia; however, when such a "different combined with other maturation which disrupt genes controlling proliferation, the result is the uncontrolled growth of an immature clone of cells, leading to the clinical entity of AML.

27
Clinical feature. The symptoms of AML are caused by replacement of normal bone marrow with leukemic cells, which causes a drop in red blood cells, platelets, and normal white blood cells. These symptoms include fatigue, shortness of breath, easy bruising and bleeding, and increased risk of infection

28
M0 Undifferentiated acute myeloblastic leukemia.
The classification of AML FAB classification. M0 Undifferentiated acute myeloblastic leukemia. M1 Acute myeloblastic leukemia with minimal maturation. M2 Acute myeloblastic leukemia with maturation. M3 Acute promyelocytic leukemia. M4 Acute myelomonocytic leukemia. M4 eosAcute myelomonocytic leukemia with eosinophilia. Acute monocytic leukemia. M6 Acute erythroid leukemia. M7 Acute megakaryoblastic leukemia.

29
Criteria for the diagnosis of acute myeloid leukaemia of M0
Blasts .30% of bone marrow nucleated cells Blasts .30% of bone marrow non-erythroid cells <3% of blasts positive for Sudan black B or for myeloperoxidase by light microscopy. Blasts demonstrated to be myeloblasts by immunological markers or by ultrastructural cytochemistry.

30
AML M0 without differentiation

31
Criteria for the diagnosis of acute myeloid leukaemia of M1.
Blasts 30% of bone marrow cells .Blasts .90% of bone marrow non-erythroid cells .3% of blasts positive for peroxidase or Sudan black B Bone marrow maturing monocytic component (promonocytes to monocytes) .10% of non-erythroid cells Bone marrow maturing granulocytic component (promyelocytes to polymorphonuclear leucocytes) .10% of non-erythroid cells
 .10% of non-erythroid cells. Bone marrow maturing granulocytic component (promyelocytes to. polymorphonuclear leucocytes) .10% of non-erythroid cells.")
32
AMLM1 with minimal maturation.

33
Criteria for the diagnosis of acute myeloid leukaemia of M2.
Blasts 30% of bone marrow cells. Blasts 30–89% of bone marrow non-erythroid cells Bone marrow maturing granulocytic component (promyelocytes to polymorphonuclear leucocytes) >10% of non-erythroid cells Bone marrow monocytic component (monoblasts to monocytes) <20% of non-erythroid cells and other criteria for M4 not met
 >10% of non-erythroid cells. Bone marrow monocytic component (monoblasts to monocytes) <20% of non-erythroid cells and other criteria for M4 not met.")
34
AML M2 shows Auer Rod and maturation

35
Acute hypergranular promyelocytic Leukaemia M3 AML
In acute hypergranular promyelocytic leukaemia the predominant cell is a highly abnormal promyelocyte. In the majority of cases, blasts are fewer than 30% of bone marrow nucleated cells. The distinctive cytological features are sufficient to permit a diagnosis and

36
In some cases there are giant granules or
multiple Auer rods, which are often present in sheaves or ‘faggots’. Most cases have a minority of cells that are agranular. M3 AML has been found to be very sensitive to the differentiating capacity of all-trans- retinoic acid (ATRA). Following such therapy an increasing proportion of cells beyond the promyelocyte stage are apparent.
. Following such therapy. an increasing proportion of cells beyond the. promyelocyte stage are apparent.")
37
AML M3 leukaemic promyelocytes

38
AML M3

39
Criteria for the diagnosis of acute myeloid leukaemia of M4.
Blasts .30% of bone marrow cells Blasts .30% of bone marrow non-erythroid cells Bone marrow granulocytic component 20% of non-erythroid cells Significant monocytic component as shown by one of the following: Bone marrow monocytic component 20% of non-erythroid cells and peripheral blood monocytic. Bone marrow resembling M2 but peripheral blood monocytic component .5000/cumm.

40
AML M4 myeloblast and leukaemic monocyte

41
Criteria for the diagnosis of acute myeloid leukaemia of M5
Blasts .30% of bone marrow cells Blasts .30% of bone marrow non-erythroid cells Bone marrow monocytic component .80% of non-erythroid cells Acute monoblastic leukaemia (M5a) Monoblasts .80% of bone marrow monocytic component Acute monocytic leukaemia (M5b) Monoblasts <80% bone marrow monocytic component
 Monoblasts .80% of bone marrow monocytic component. Acute monocytic leukaemia (M5b) Monoblasts <80% bone marrow monocytic component.")
42
AML M5 monoblasts

43
Criteria for the diagnosis of acute myeloid leukaemia of M6
Erythroblasts .50% of bone marrow nucleated cells Blasts 30% of bone marrow non-erythroid cells

44
AML M6 erythroblasts

45
Criteria for the diagnosis of acute myeloid leukaemia of M7
Blasts 30% of bone marrow nucleated cells. Blasts demonstrated to be megakaryoblasts by immunological markers, ultrastructural examination or ultrastructural cytochemistry

46
AML M7 megakaryoblast

47
AML M7 many megakaryocytes

48
The WHO classification of AML.
Therapy-related AML and MDS. Alkylating agent-related Topoisomerase II-inhibitor-related Other types AML with recurrent cytogenetic abnormalities* AML with t(8;21)(q22;q22) AML with abnormal bone marrow eosinophils with inv(16)(p13q22) or t(16;16)(p13;q22) Acute promyelocytic leukemia with t(15;17)(q22;q12) AML with 11q23 (MLL) abnormalities. AML with multilineage dysplasia following MDS. AML not otherwise categorized. This group is nearly similar to FAB group, but blast cells are 20% in stead of 30%
(q22;q22) AML with abnormal bone marrow eosinophils with. inv(16)(p13q22) or t(16;16)(p13;q22) Acute promyelocytic leukemia with. t(15;17)(q22;q12) AML with 11q23 (MLL) abnormalities. AML with multilineage dysplasia following MDS. AML not otherwise categorized. This group is nearly similar to FAB group, but blast cells are 20% in stead of 30%")
49
CHRONIC MYELOID LEUKAEMIAS

50
The World Health Organization (WHO)
classification assigns some chronic myeloid leukaemias to a myeloproliferative category and others, in which there are also dysplastic features, to a myeloproliferative/myelodysplastic category

51
Classification of the chronic myeloid leukaemias, based on the WHO classification.
Myeloproliferative disorders Chronic myelogenous leukaemia Chronic neutrophilic leukaemia Chronic eosinophilic leukaemia Basophilic leukaemia Mast cell leukaemia Myelodysplastic/myeloproliferative disorders Chronic myelomonocytic leukaemia Chronic myelomonocytic leukaemia with eosinophilia Myelodysplastic/myeloproliferative disorder associated with t(5;12)(q33;p13)* Atypical chronic myeloid leukaemia Juvenile myelomonocytic leukaemia
(q33;p13)* Atypical chronic myeloid leukaemia. Juvenile myelomonocytic leukaemia.")
52
Chronic granulocytic leukaemia
Chronic granulocytic leukaemia (CGL) is a disease entity with specific haematological, cytogenetic and molecular genetic features. Alternative designations are chronic myelogenous leukaemia, chronic myeloid leukaemia and chronic myelocytic leukaemia.
 is a disease. entity with specific haematological, cytogenetic. and molecular genetic features. Alternative designations are chronic myelogenous. leukaemia, chronic myeloid leukaemia and chronic. myelocytic leukaemia.")
53
CGL is a disease of bi- or triphasic with a chronic and an acute phase and, sometimes, an intervening accelerated phase

54
The chronic phase of chronic granulocytic leukaemia
Clinical and haematological features. CGL is predominantly a disease of adults. The usual clinical presentation is with splenomegaly, hepatomegaly, symptoms of anaemia, and systemic symptoms such as sweating and weight loss. Occasionally this is an incidental diagnosis when a blood count is performed for another reason.

55
The peripheral blood usually shows anemia
and leucocytosis with a very characteristic differential count. The two predominant cell types are the myelocyte and the mature neutrophil .

56
Almost all patients have an absolute
basophilia and more than 90% have eosinophilia. The platelet count is most often normal or somewhat elevated but is low in about 5% of cases.

57
BM film of a patient with CGL showing neutrophil leucocytosis with left shift.

58
The bone marrow is intensely hypercellular
with marked granulocytic hyperplasia and with the myeloid/erythroid (M:E) ratio being greater than 10:1. There is hyperplasia of neutrophil, eosinophil and basophil lineages.
 ratio. being greater than 10:1. There is hyperplasia of neutrophil, eosinophil and basophil lineages.")
59
Bone marrow shows myeloid hyperplasia.

60
CGL in accelerated phase and blast Transformation
After a variable period in chronic phase, usually several years, CGL undergoes further evolution. There may be an abrupt transformation to an Acute leukaemia, designated blast transformation, or there may be an intervening phase of accelerated disease.

61
The WHO group have suggested the following criteria for accelerated phase:
(i) Myeloblasts constitute 10–19% of peripheral blood white cells or bone marrow nucleated cells. (ii) peripheral blood basophiles are 20% or more of nucleated cells. (iii) there is persistent thrombocytopenia or persistent thrombocytosis that does not respond to treatment. (iv) there is an increasing white cell count and increasing spleen size that does not respond to treatment. (v) cytogenetic evolution . (vi) there is marked granulocyte dysplasia or prominent proliferation of small dysplastic megakaryocytes in large clusters or sheets.
 Myeloblasts constitute 10–19% of peripheral blood. white cells or bone marrow nucleated cells. (ii) peripheral blood basophiles are 20% or more of. nucleated cells. (iii) there is persistent thrombocytopenia or persistent. thrombocytosis that does not respond to treatment. (iv) there is an increasing white cell count and increasing. spleen size that does not respond to treatment. (v) cytogenetic evolution . (vi) there is marked granulocyte dysplasia or prominent. proliferation of small dysplastic megakaryocytes in. large clusters or sheets.")
62
Transformation may be myeloid or lymphoid.
Blast transformation phase. Transformation may be myeloid or lymphoid. It is important to make the distinction since there lymphoblastic transformation. Lymphoid blast crisis is more likely to emerge suddenly without a preceding accelerated phase

63
Cytogenetic and molecular genetic features
CGL was the first malignant disease for which a consistent association with an acquired non- random cytogenetic abnormality was recognized. In 1960 Nowell and Hungerford reported its Association with an abnormal chromosome designated the Philadelphia (Ph) chromosome after the city of its discovery.
 chromosome after the city of its discovery.")
64
Karyotype of a patient with CGL showing t(9;22)
")
65
Chronic lymphocytic leukaemia

66
Chronic lymphocytic leukaemia (CLL) is a chronic
B-lineage lymphoproliferative disorder defined by characteristic morphology and immunophenotype. Small lymphocytic lymphoma is an equivalent lymphoma without circulating neoplastic cells

67
CLL is the most common leukaemia in western
Europe and North America with an incidence in different surveys varying between 1 and more than 10/ /year. The incidence is lower in Chinese, Japanese and South American Indians. It is typically a disease of the elderly with a higher incidence in males

68
Clinical feature. In the later stages, CLL is characterized
by lymphadenopathy, hepatomegaly, splenomegaly and eventually by impairment of bone marrow function. In the early stages of the disease there are no symptoms or abnormal physical findings and the diagnosis is made incidentally

69
Various arbitrary levels of absolute lymphocyte
count have been suggested for the diagnosis of CLL (for example greater than /cumm). But the demonstration of a monoclonal population of B lymphocytes with a characteristic immunophenotype permits diagnosis at an earlier stage when the lymphocyte count is less elevated.
. But the demonstration of a monoclonal population. of B lymphocytes with a characteristic. immunophenotype permits diagnosis at an earlier. stage when the lymphocyte count is less elevated.")
70
A scoring system for the immunophenotypic diagnosis of chronic lymphocytic leukaemia (CLL)
Score 1 for each of the following: • Weak expression of SmIg • Expression of CD5 • Expression of CD23 • No expression of FMC7 • No expression of CD22 A score of ≥4 points is confirmatory of CLL

71
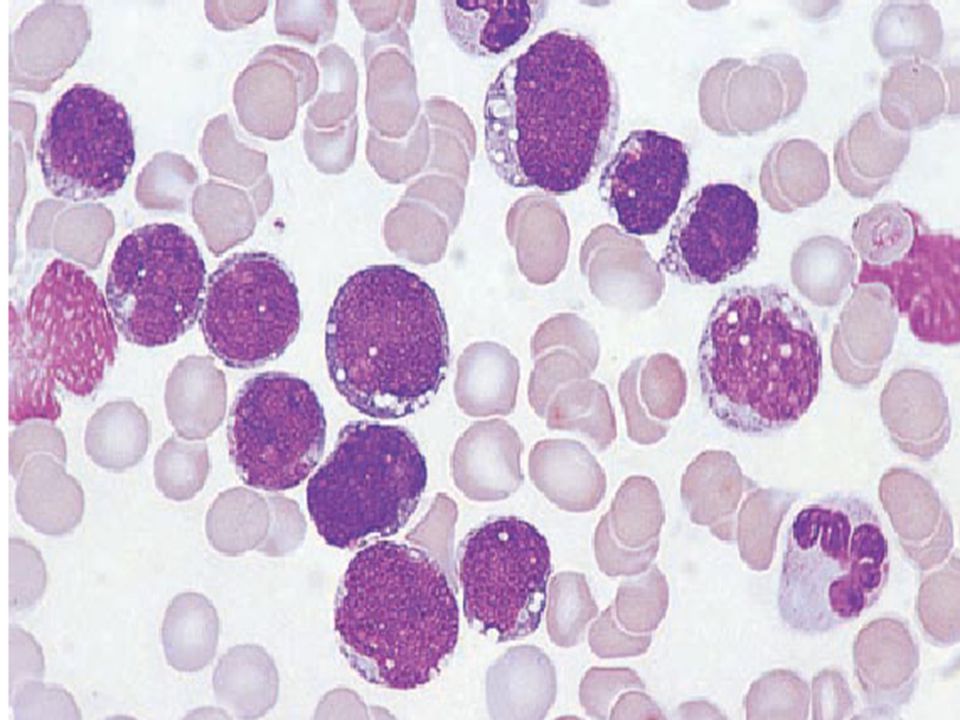
Peripheral blood chronic lymphocytic leukaemia showing
two mature lymphocytes and one smear cell

72
Peripheral blood findings
In the early stages of the disease the Peripheral blood abnormality is confined to the lymphocytes. Later in the disease course there is a normocytic, normochromic anaemia and thrombocytopenia. Neutropenia is uncommon unless cytotoxic therapy has been administered

73
Bone marrow findings. The bone marrow aspirate is hypercellular
as a consequence of infiltration by lymphocytes with similar features to those in the peripheral blood. Lymphocytes percentage in the bone marrow is 40% of all nucleated marrow cells total.

74
Rai staging system for chronic lymphocytic leukaemia
0 Peripheral blood and bone marrow lymphocytosis only. I Intermediate Lymphocytosis and lymphadenopathy. II Intermediate Lymphocytosis plus hepatomegaly, splenomegaly or both. III Lymphocytosis and anaemia (haemoglobin concentration less than 11 g/dl). IV Lymphocytosis and thrombocytopenia (platelet count less than /cmm)
. IV Lymphocytosis and thrombocytopenia (platelet count. less than /cmm)")
75
CLL Transformation. Chronic lymphocytic leukaemia may undergo
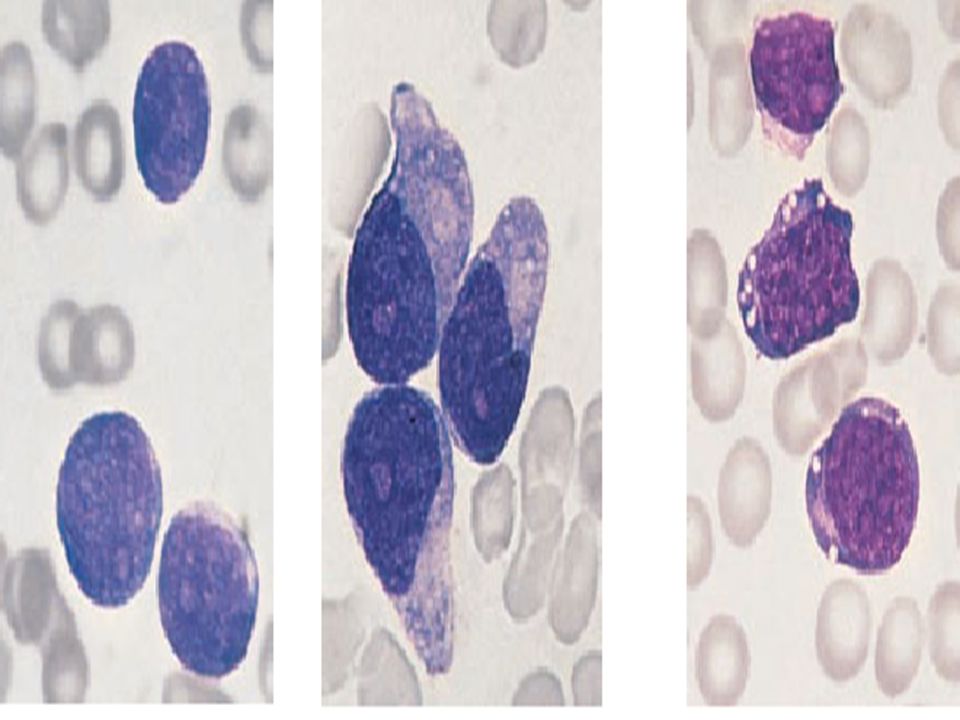
two types of transformation. 1-Prolymphocytoid transformation. 2-large cell transformation, referred to as Richter’s syndrome.

76
CLL with transformation to PLL

Similar presentations



1 CHILDHOOD LEUKAEMIA. TA OGUNLESI (FWACP)2 LEUKAEMIA Heterogenous group of malignant disorders Characterised by uncontrolled clonal.>")










 Myelodysplastic / myeloproliferative diseases (MDS/MPD) >")

:>")
 leukemia Is characterized by an unregulated proliferation of myeloid elements in the bone marrow,>")


 to those.>")
>")
