Download presentation
Presentation is loading. Please wait.

1
Andrzej Kolinski LABORATORY OF THEORY OF BIOPOLYMERS WARSAW UNIVERSITY http://www.biocomp.chem.uw.edu.pl Structure and Function of Biomolecules, Bedlewo, May 12-15, 2004 Andrzej Kolinski LABORATORY OF THEORY OF BIOPOLYMERS WARSAW UNIVERSITY http://www.biocomp.chem.uw.edu.pl Structure and Function of Biomolecules, Bedlewo, May 12-15, 2004 HIGH RESOLUTION LATTICE MODELS OF PROTEINS: DESIGN & APPLICATIONS

2
WHY REDUCED MODELS? Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - “long” MD simulations cover 100 nanoseconds). Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - “long” MD simulations cover 100 nanoseconds). The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - “long” MD simulations cover 100 nanoseconds). Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - “long” MD simulations cover 100 nanoseconds). The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases.
. Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - long MD simulations cover 100 nanoseconds). The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - long MD simulations cover 100 nanoseconds). Classical Molecular Mechanics study of the large scale conformational rearrangements of biomolecules are still impractical (proteins fold in a time frame of 0.001s to 100s - long MD simulations cover 100 nanoseconds). The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. The number of degrees of freedom treated in an explicit way needs to be reduced and the energy landscape smoothened. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. Knowledge-based force fields of reduced models seem to have frequently a higher predictive power than the all-atom potentials of the Molecular Mechanics. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases. We know about 1000 times more protein sequences than protein structures (ca. 30M against ca. 30k). This gap increases..")
3
OUTLINEOUTLINE Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Ab initio folding (an illustration) Ab initio folding (an illustration) Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Comparative modeling starting from multiple threading alignments Comparative modeling starting from multiple threading alignments Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Ab initio folding (an illustration) Ab initio folding (an illustration) Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Comparative modeling starting from multiple threading alignments Comparative modeling starting from multiple threading alignments
 modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Comparative modeling starting from multiple threading alignments Comparative modeling starting from multiple threading alignments Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Reduced protein models of an intermediate and high resolution (representation, sampling and force field) Ab initio folding (an illustration) Ab initio folding (an illustration) Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Loops (or fragments) modeling using various reduced representations: SICHO, CABS and REFINER models. Comparison with standard modeling tools: MODELLER and SWISS-MODEL Comparative modeling starting from multiple threading alignments Comparative modeling starting from multiple threading alignments.")
4
SICHO, CABS and REFINER All models use knowledge-based statistical potentials derived via an analysis of structural regularities seen in the solved structures of globular proteins All models use knowledge-based statistical potentials derived via an analysis of structural regularities seen in the solved structures of globular proteins

5
Sampling of the conformational space of the SICHO and CABS models - Single residue moves -Two-residue moves -Three-residue moves -Small distance (rigid body) moves of a randomly selected fragment of the a randomly selected fragment of the model chain model chain -Reptation type moves - Single residue moves -Two-residue moves -Three-residue moves -Small distance (rigid body) moves of a randomly selected fragment of the a randomly selected fragment of the model chain model chain -Reptation type moves
 moves of a randomly selected fragment of the a randomly selected fragment of the model chain model chain -Reptation type moves - Single residue moves -Two-residue moves -Three-residue moves -Small distance (rigid body) moves of a randomly selected fragment of the a randomly selected fragment of the model chain model chain -Reptation type moves")
6
Conformational Search Scheme

7
INTERACTION SCHEME Generic “protein-like” biases Generic “protein-like” biases Statistical potentials for short-range conformational propensities Statistical potentials for short-range conformational propensities Model of main chain hydrogen bonds Model of main chain hydrogen bonds Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials) Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials) Generic “protein-like” biases Generic “protein-like” biases Statistical potentials for short-range conformational propensities Statistical potentials for short-range conformational propensities Model of main chain hydrogen bonds Model of main chain hydrogen bonds Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials) Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials)
 Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials) Generic protein-like biases Generic protein-like biases Statistical potentials for short-range conformational propensities Statistical potentials for short-range conformational propensities Model of main chain hydrogen bonds Model of main chain hydrogen bonds Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials) Pairwise interactions between united atoms (including orientation- and secondary structure dependent potentials)")
8
Generic (sequence independent) chain stiffness - regular secondary structure propensities
 chain stiffness - regular secondary structure propensities")
9
Generic (sequence independent) chain stiffness 1 B1 = f × g for: (v i-1 v i+3 )<0 B1 = f × g for: (v i-1 v i+3 )<0 B2 = -f × g -g × g for: | r i+4 –r i |< 7.0 Å and “right handed” twist or: | r i+4 –r i |>11.0 Å and -type geometry B2 = -f × g -g × g for: | r i+4 –r i |< 7.0 Å and “right handed” twist or: | r i+4 –r i |>11.0 Å and -type geometry
 chain stiffness 1 B1 = f × g for: (v i-1 v i+3 )<0 B1 = f × g for: (v i-1 v i+3 )<0 B2 = -f × g -g × g for: | r i+4 –r i |< 7.0 Å and right handed twist or: | r i+4 –r i |>11.0 Å and -type geometry B2 = -f × g -g × g for: | r i+4 –r i |< 7.0 Å and right handed twist or: | r i+4 –r i |>11.0 Å and -type geometry")
10
Generic (sequence independent) chain stiffness 1 B4 = h × g for: (r i+5 –r i ) (r i+ 10 –r i+5 ) 0 i.e., penalty for a too crumpled main chain conformations B4 = h × g for: (r i+5 –r i ) (r i+ 10 –r i+5 ) 0 i.e., penalty for a too crumpled main chain conformations For known or strongly predicted secondary structure fragments an additional bias towards proper values of the medium-range distances along the chain could be superimposed
 chain stiffness 1 B4 = h × g for: (r i+5 –r i ) (r i+ 10 –r i+5 ) 0 i.e., penalty for a too crumpled main chain conformations B4 = h × g for: (r i+5 –r i ) (r i+ 10 –r i+5 ) 0 i.e., penalty for a too crumpled main chain conformations For known or strongly predicted secondary structure fragments an additional bias towards proper values of the medium-range distances along the chain could be superimposed")
11
Short-range conformational propensities E 13 (r i+2,i, A i, A i+2 ) E 14 (r* i+3,i, A i+1, A i+2 E 15 (r i+4,i, A i+1, A i+3 ) Note: the reduced backbone geometry correlates better with secondary structure than the phi-psi angles E 13 (r i+2,i, A i, A i+2 ) E 14 (r* i+3,i, A i+1, A i+2 E 15 (r i+4,i, A i+1, A i+3 ) Note: the reduced backbone geometry correlates better with secondary structure than the phi-psi angles -10-1 0 1 10 _______________________________________________________________________________________________________ ALA -0.25 -0.45 -0.39 0.73 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 -1.12 -2.55 0.44 0.56 0.25 0.76 0.51 VAL THR -1.71 -1.83 0.06 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 0.11 -1.51 0.56 0.56 0.44 -0.57 -0.75 _______________________________________________________________________________________________________ Left-handed beta unlike or prohibited Alpha Right-handed beta -10-1 0 1 10 _______________________________________________________________________________________________________ ALA -0.25 -0.45 -0.39 0.73 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 -1.12 -2.55 0.44 0.56 0.25 0.76 0.51 VAL THR -1.71 -1.83 0.06 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 2.00 0.11 -1.51 0.56 0.56 0.44 -0.57 -0.75 _______________________________________________________________________________________________________ Left-handed beta unlike or prohibited Alpha Right-handed beta E/kT ~ -ln (n k,A1,A2 / n k,Ai,Aj >) average over the database E/kT ~ -ln (n k,A1,A2 / n k,Ai,Aj >) average over the database
 E 14 (r* i+3,i, A i+1, A i+2 E 15 (r i+4,i, A i+1, A i+3 ) Note: the reduced backbone geometry correlates better with secondary structure than the phi-psi angles E 13 (r i+2,i, A i, A i+2 ) E 14 (r* i+3,i, A i+1, A i+2 E 15 (r i+4,i, A i+1, A i+3 ) Note: the reduced backbone geometry correlates better with secondary structure than the phi-psi angles _______________________________________________________________________________________________________ ALA VAL THR _______________________________________________________________________________________________________ Left-handed beta unlike or prohibited Alpha Right-handed beta _______________________________________________________________________________________________________ ALA VAL THR _______________________________________________________________________________________________________ Left-handed beta unlike or prohibited Alpha Right-handed beta E/kT ~ -ln (n k,A1,A2 / n k,Ai,Aj >) average over the database E/kT ~ -ln (n k,A1,A2 / n k,Ai,Aj >) average over the database")
12
CABS reduced representation

13
Model of the main chain hydrogen bonds Hydrogen bonds cause specific spatial arrangement of the -trace vectors and the -carbon united atoms The united atoms i and j are “hydrogen bonded” when: - at least one of the vectors h points into the vicinity of the -carbon i or j - vectors h are “almost” parallel (or antiparallel) - (bi * bj) >0 (“roughly” parallel) The strength of the hydrogen bond is moderated by a cooperative component dependent on the distance between the corresponding centers of the C -C virtual bonds (minimum of the potential at 4.25 Å ) The united atoms i and j are “hydrogen bonded” when: - at least one of the vectors h points into the vicinity of the -carbon i or j - vectors h are “almost” parallel (or antiparallel) - (bi * bj) >0 (“roughly” parallel) The strength of the hydrogen bond is moderated by a cooperative component dependent on the distance between the corresponding centers of the C -C virtual bonds (minimum of the potential at 4.25 Å ) Additional rules: No hydrogen bonds between pairs assigned as (HE) and (HH for |i-j|>3) The C -based model of hydrogen bonds correlates very well with the real hydrogen bonds. When “translating” the indices need to be properly shifted (by +/- 1) depending on type of secondary structure Additional rules: No hydrogen bonds between pairs assigned as (HE) and (HH for |i-j|>3) The C -based model of hydrogen bonds correlates very well with the real hydrogen bonds. When “translating” the indices need to be properly shifted (by +/- 1) depending on type of secondary structure
 depending on type of secondary structure Additional rules: No hydrogen bonds between pairs assigned as (HE) and (HH for |i-j|>3) The C -based model of hydrogen bonds correlates very well with the real hydrogen bonds. When translating the indices need to be properly shifted (by +/- 1) depending on type of secondary structure.")
14
Pairwise interactions (C , C , Side Groups) Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Soft core excluded volume for interactions with the side groups. Soft core excluded volume for interactions with the side groups. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Two side groups are assumed to be “in contact” when any pair of their heavy atoms is “in contact” (4.5 cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Two side groups are assumed to be “in contact” when any pair of their heavy atoms is “in contact” (4.5 Å cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Side group pairwise potentials are “context” dependent (mutual orientation, conformation of the main chain) Side group pairwise potentials are “context” dependent (mutual orientation, conformation of the main chain) Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Soft core excluded volume for interactions with the side groups. Soft core excluded volume for interactions with the side groups. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Two side groups are assumed to be “in contact” when any pair of their heavy atoms is “in contact” (4.5 cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Two side groups are assumed to be “in contact” when any pair of their heavy atoms is “in contact” (4.5 Å cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Side group pairwise potentials are “context” dependent (mutual orientation, conformation of the main chain) Side group pairwise potentials are “context” dependent (mutual orientation, conformation of the main chain)
. Soft core excluded volume for interactions with the side groups. Soft core excluded volume for interactions with the side groups. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Two side groups are assumed to be in contact when any pair of their heavy atoms is in contact (4.5 cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Two side groups are assumed to be in contact when any pair of their heavy atoms is in contact (4.5 Å cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Side group pairwise potentials are context dependent (mutual orientation, conformation of the main chain) Side group pairwise potentials are context dependent (mutual orientation, conformation of the main chain) Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Hard-core excluded volume for C -C , C -C and C -C pairs (the cut-off distances are amino acid independent). Soft core excluded volume for interactions with the side groups. Soft core excluded volume for interactions with the side groups. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Pairwise potentials for side groups derived from a statistical analysis of known protein structures. Two side groups are assumed to be in contact when any pair of their heavy atoms is in contact (4.5 cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Two side groups are assumed to be in contact when any pair of their heavy atoms is in contact (4.5 Å cut- off) – the average distance between the centers of mass are then taken as a contact distance for a pair of side groups. Side group pairwise potentials are context dependent (mutual orientation, conformation of the main chain) Side group pairwise potentials are context dependent (mutual orientation, conformation of the main chain).")
15
Pairwise interactions of the side groups Between centers of mass (all heavy atoms of a side group + C ). Cut-off distances pairwise dependent (not additive, account for some packing details). Square-well shape of the potential (for charged residues a tail added). Soft (however relatively large) excluded volume potential – the height is amino acid independent. For a given pair of amino acids the strength of interactions and the cut-off distances depend on mutual orientation of the interacting side groups and on the local geometry of the main chain. Between centers of mass (all heavy atoms of a side group + C ). Cut-off distances pairwise dependent (not additive, account for some packing details). Square-well shape of the potential (for charged residues a tail added). Soft (however relatively large) excluded volume potential – the height is amino acid independent. For a given pair of amino acids the strength of interactions and the cut-off distances depend on mutual orientation of the interacting side groups and on the local geometry of the main chain.
. Square-well shape of the potential (for charged residues a tail added). Soft (however relatively large) excluded volume potential – the height is amino acid independent. For a given pair of amino acids the strength of interactions and the cut-off distances depend on mutual orientation of the interacting side groups and on the local geometry of the main chain. Between centers of mass (all heavy atoms of a side group + C ). Cut-off distances pairwise dependent (not additive, account for some packing details). Square-well shape of the potential (for charged residues a tail added). Soft (however relatively large) excluded volume potential – the height is amino acid independent. For a given pair of amino acids the strength of interactions and the cut-off distances depend on mutual orientation of the interacting side groups and on the local geometry of the main chain..")
16
CONTEXT-DEPENDENT STATISTICAL POTENTIALS Three types of the mutual orientations of the side groups: A-antiparallel, M-intermediate, P-parallel Two types of the main chain conformations: C- compact and E-extended Two types of the main chain conformations: C- compact and E-extended Derived pairwise contact potentials from the statistics of the numbers of parallel, antiparllel and semi-orthogonal contacts for a given residue type and two types of the main chain conformations.

17
NEW STATISTICAL POTENTIALS (AN EXAMPLE) LYS-GLU POTENTIAL PMA CC -0.9 -0.4 0.9 EE -1.1 -0.4 0.6 CE -0.2 0.1 0.8 EC -0.2 0.0 0.8 LYS-GLU POTENTIAL PMA CC -0.9 -0.4 0.9 EE -1.1 -0.4 0.6 CE -0.2 0.1 0.8 EC -0.2 0.0 0.8 GAPLESS THREADING %NATIVE Z-score QUASI 86 % 6.72 QUASI3 94 % 7.84 QUASI3S 97 % 9.96 When tested on a large set of decoys the orientation and backbone conformation dependent potentials QUASI3S exhibits better correlation between energy and RMSD from native than the more “generic” potentials
 LYS-GLU POTENTIAL PMA CC EE CE EC LYS-GLU POTENTIAL PMA CC EE CE EC GAPLESS THREADING %NATIVE Z-score QUASI 86 % 6.72 QUASI3 94 % 7.84 QUASI3S 97 % 9.96 When tested on a large set of decoys the orientation and backbone conformation dependent potentials QUASI3S exhibits better correlation between energy and RMSD from native than the more generic potentials")
18
Ab initio folding “Pure” ab initio (with only statistical potentials) protein folding and macromolecular assembly (results for the SICHO model) “Pure” ab initio (with only statistical potentials) protein folding and macromolecular assembly (results for the SICHO model)
 protein folding and macromolecular assembly (results for the SICHO model) Pure ab initio (with only statistical potentials) protein folding and macromolecular assembly (results for the SICHO model)")
21
LOOP MODELING – STRUCTURE COMPLETION Fixed template (and an “ideal” alignment) from PDB with removed fragments of their native structure Fixed template (and an “ideal” alignment) from PDB with removed fragments of their native structure Random starting conformation of the loops (non- entangled) Random starting conformation of the loops (non- entangled) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) No human intervention during the modeling procedures No human intervention during the modeling procedures Fixed template (and an “ideal” alignment) from PDB with removed fragments of their native structure Fixed template (and an “ideal” alignment) from PDB with removed fragments of their native structure Random starting conformation of the loops (non- entangled) Random starting conformation of the loops (non- entangled) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) No human intervention during the modeling procedures No human intervention during the modeling procedures
 from PDB with removed fragments of their native structure Fixed template (and an ideal alignment) from PDB with removed fragments of their native structure Random starting conformation of the loops (non- entangled) Random starting conformation of the loops (non- entangled) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) No human intervention during the modeling procedures No human intervention during the modeling procedures Fixed template (and an ideal alignment) from PDB with removed fragments of their native structure Fixed template (and an ideal alignment) from PDB with removed fragments of their native structure Random starting conformation of the loops (non- entangled) Random starting conformation of the loops (non- entangled) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) Loop optimization using SICHO, CABS and REFINER (sampling via Replica Exchange Monte Carlo) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) The lowest energy structure taken for a comparison with MODELLER and SWISS-MODEL (automatic version) No human intervention during the modeling procedures No human intervention during the modeling procedures")
22
EXAMPLES (a-SICHO, b-CABS, c-REFINER, d-MODELLER) Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment
 Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment")
23
EXAMPLES (a-SICHO, b-CABS, c-REFINER, d-MODELLER) Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template
 Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template Green – native fragment or loop removed from the PDB structure Red – Modeled fragment Gray – template")
24
COMPARATIVE MODELING WITH MULTIPLE TEMPLATES Highest score templates detected by threading procedures are used to extract the distance restraints Highest score templates detected by threading procedures are used to extract the distance restraints “Soft” implementation of the restraints in the CABS algorithm (from the top-four templates – when available) “Soft” implementation of the restraints in the CABS algorithm (from the top-four templates – when available) Sampling via Replica Exchange Monte Carlo Sampling via Replica Exchange Monte Carlo Almost always a single cluster of structures is obtained and its centroid is taken as a final model Almost always a single cluster of structures is obtained and its centroid is taken as a final model Highest score templates detected by threading procedures are used to extract the distance restraints Highest score templates detected by threading procedures are used to extract the distance restraints “Soft” implementation of the restraints in the CABS algorithm (from the top-four templates – when available) “Soft” implementation of the restraints in the CABS algorithm (from the top-four templates – when available) Sampling via Replica Exchange Monte Carlo Sampling via Replica Exchange Monte Carlo Almost always a single cluster of structures is obtained and its centroid is taken as a final model Almost always a single cluster of structures is obtained and its centroid is taken as a final model
 Soft implementation of the restraints in the CABS algorithm (from the top-four templates – when available) Sampling via Replica Exchange Monte Carlo Sampling via Replica Exchange Monte Carlo Almost always a single cluster of structures is obtained and its centroid is taken as a final model Almost always a single cluster of structures is obtained and its centroid is taken as a final model Highest score templates detected by threading procedures are used to extract the distance restraints Highest score templates detected by threading procedures are used to extract the distance restraints Soft implementation of the restraints in the CABS algorithm (from the top-four templates – when available) Soft implementation of the restraints in the CABS algorithm (from the top-four templates – when available) Sampling via Replica Exchange Monte Carlo Sampling via Replica Exchange Monte Carlo Almost always a single cluster of structures is obtained and its centroid is taken as a final model Almost always a single cluster of structures is obtained and its centroid is taken as a final model")
25
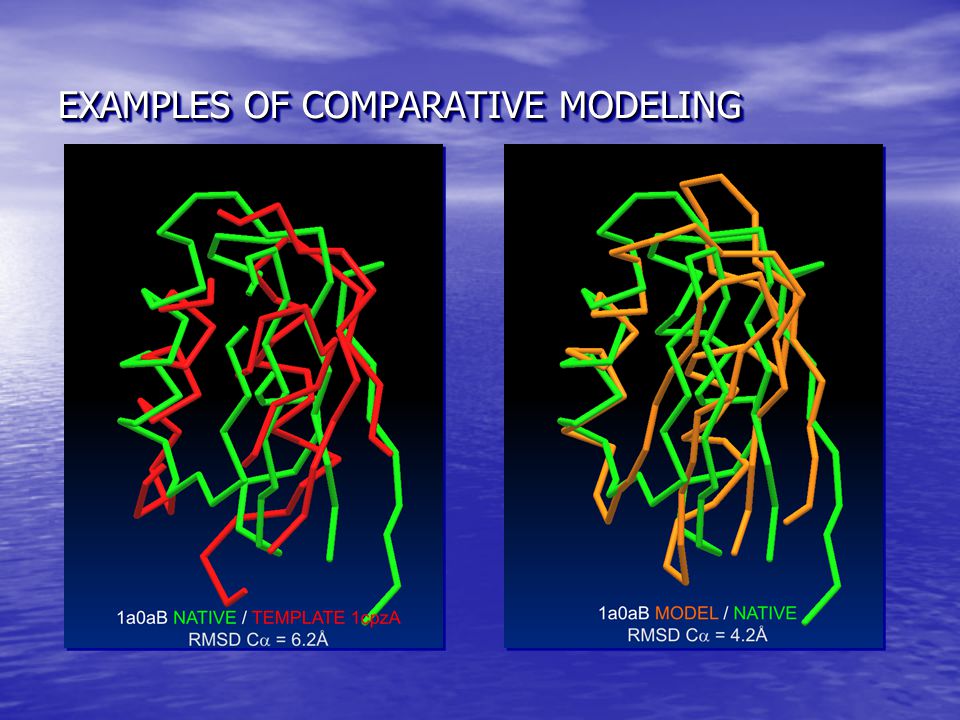
EXAMPLES OF COMPARATIVE MODELING

27
SUMMARY OF COMPARATIVE MODELING SUMMARY OF COMPARATIVE MODELING Frequently the models are closer to the native structure than to any of the templates

28
CONCLUSIONSCONCLUSIONS Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Applications: Applications: - ab initio structure prediction - ab initio structure prediction - comparative modeling (also multitemplate) - comparative modeling (also multitemplate) - structure assembly from sparse experimental data - structure assembly from sparse experimental data - dynamics and thermodynamics of proteins, prions - dynamics and thermodynamics of proteins, prions - flexible docking, macromolecular assemblies - flexible docking, macromolecular assemblies Tools exist for the all-atom reconstruction of the reduced models. (See: Tools exist for the all-atom reconstruction of the reduced models. (See: NIH Research Resources for Multiscale Modeling Tools in Structural Biology hhtp://mmtsb.scripps.edu) Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Applications: Applications: - ab initio structure prediction - ab initio structure prediction - comparative modeling (also multitemplate) - comparative modeling (also multitemplate) - structure assembly from sparse experimental data - structure assembly from sparse experimental data - dynamics and thermodynamics of proteins, prions - dynamics and thermodynamics of proteins, prions - flexible docking, macromolecular assemblies - flexible docking, macromolecular assemblies Tools exist for the all-atom reconstruction of the reduced models. (See: Tools exist for the all-atom reconstruction of the reduced models. (See: NIH Research Resources for Multiscale Modeling Tools in Structural Biology hhtp://mmtsb.scripps.edu)
 Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Algorithms employing reduced representation of the protein conformational space are now mature and efficient tools for protein modeling Applications: Applications: - ab initio structure prediction - ab initio structure prediction - comparative modeling (also multitemplate) - comparative modeling (also multitemplate) - structure assembly from sparse experimental data - structure assembly from sparse experimental data - dynamics and thermodynamics of proteins, prions - dynamics and thermodynamics of proteins, prions - flexible docking, macromolecular assemblies - flexible docking, macromolecular assemblies Tools exist for the all-atom reconstruction of the reduced models. (See: Tools exist for the all-atom reconstruction of the reduced models. (See: NIH Research Resources for Multiscale Modeling Tools in Structural Biology hhtp://mmtsb.scripps.edu).")
29
Acknowledgement Warsaw University Warsaw University Poland Poland Michal Boniecki Dominik Gront Sebastian Kmiecik Sebastian Kmiecik Piotr Klein Piotr Pokarowski Piotr Rotkiewicz Andrzej Kolinski Andrzej Kolinski SUNY at Buffalo (NY) SUNY at Buffalo (NY) Piotr Rotkiewicz Jeffrey Skolnick More info: http://www.biocomp.chem.uw.edu.pl
 SUNY at Buffalo (NY) Piotr Rotkiewicz Jeffrey Skolnick More info:")
Similar presentations














 threading loop building re-packing side-chains in core, DEE, SCWRL.>")
![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")

 Today: Why bother about proteins/prediction Concepts of molecular.>")



