Download presentation
Presentation is loading. Please wait.

2
Crystals are made from very large numbers of small units: Unit cells. The unit cell may contain more than one protein. The packing of the unit cells gives rise to a 3D structure with clear symmetry: a protein crystal.

3
Protein crystals have different symmetry forms. They are hardly visible by eye.

4
Good crystals diffract X-ray radiation (wavelength in the order of 0.5 tot 2.0 Angstrom) very well in a regular pattern. In the diffraction pattern (b) more than 100.000 diffracted beams are collected.
 more than diffracted beams are collected..")
5
Protein crystals contain large channels and holes filled with solvent. Usually around 50% of the total volume is solvent.

6
The usual method to obtain protein crystals is via the hanging drop method. About 10 l of a 10 mg/ml protein solution is put on a thin glass plate. In the precipitant solution the concentration of ammonium sulfate (the most frequently used salt) is higher than in the protein solution. Water evaporated from the top is taken up by the precipitant. After some time (weeks) the proteins in the solution start to crystallize (depending on temperature, buffer pH).
 is higher than in the protein solution. Water evaporated from the top is taken up by the precipitant. After some time (weeks) the proteins in the solution start to crystallize (depending on temperature, buffer pH)..")
7
A narrow beam of X-rays (generated via a rotating anode (university based) or via a synchroton (national or European facilities)) hits the crystal and diffract when electron density is encountered.
 or via a synchroton (national or European facilities)) hits the crystal and diffract when electron density is encountered.")
8
Most of the primary beam, when hitting a protein crystal, passes straight through it. Some X-ray interact with electrons on each atom and the x-ray beam is diffracted. Because of the symmetry in a crystal the diffracted beams interfere with one another. The relationship between the distance between the planes d and the wavelength is given by Bragg’s law: 2d sin =. This relationship enables one to determine the size of the unit cell.

9
The reflection angle for a diffracted beam can be calculated from the distance r between the diffracted spot and the position where the primary beam hits the film. From the geometry the Tangent of the angle 2 = r/A. A is the distance between crystal and film.

10
Two diffracted beams are shown. These are defined by amplitude ( this is the strength/ intensity of the beam, the higher the amplitude the more signal), the phase (this is related to the interference, negative/positive, with other beams) and wavelength (which is determined by the x-ray source). The better and intenser the monochromatic light the higher the quality of the diffraction pattern. The best quality at the moment is obtained using the synchotron facility in Grenoble, with a good second best EMBL Hamburg. The wavelength and the amplitude of the beams can be measured but the phase is a difficult problem which can usually not be solved directly from the measurements.
, the phase (this is related to the interference, negative/positive, with other beams) and wavelength (which is determined by the x-ray source). The better and intenser the monochromatic light the higher the quality of the diffraction pattern. The best quality at the moment is obtained using the synchotron facility in Grenoble, with a good second best EMBL Hamburg. The wavelength and the amplitude of the beams can be measured but the phase is a difficult problem which can usually not be solved directly from the measurements..")
11
A general method has been introduced to solve the phase problem: multiple isomorphous replacement (MIR). This requires the introduction of new x-ray scatterers in the unit cell. These additions are heavy atoms, there need to be only a very few and they should not disturb the 3D structure. With some luck p.e. SH groups exposed in the solvent channels bind heavy metals. Because heavy atoms contain many electrons they diffract the x-ray beams more strongly than H,C,N,O or S.

12
The diffraction patterns of DNA (A-DNA left) and B-DNA (right)
 and B-DNA (right)")
14
The amplitudes and phases are used to calculate an electron-density map. This map has to be fitted with the amino acid sequence. The interpretation of the map is complicated by limitations. First, the map contains errors, usually due to errors in phase angles. In addition, the quality of the map depends on the resolution of the diffracted data, and this depends on the quality of the crystal. Resolution is measured in Angstroms, the smaller this number, the better. At 2.0 Angstrom resolution the difference between sidechains are visible, but only at very high resolution (1.0 Angstrom or better) we can directly determine what amino acid is observed in the diffraction pattern.
 we can directly determine what amino acid is observed in the diffraction pattern..")
16
One dimensional 1H-NMR spectra. a)Ethanol. The chemical shifts are distinct for the different hydrogen atoms. Resonances are split into three or four peaks due to their neighbours (CH2 by the CH3 group and vise versa). b) 1H-NMR spectrum of 36 amino acid protein.
. b) 1H-NMR spectrum of 36 amino acid protein..")
17
The overlap in the 1D NMR spectrum is diminished using 2D NMR spectroscopy. The peaks on the diagonal from upper right to lower left are identical to the 1D spectrum in the previous slide. The off-diagional peaks represent through-space interaction between hydrogen atoms.

18
The interaction between atoms can be measured using different NMR methods: Through-bonds: COSY (a) Through-space: NOESY (b)
 Through-space: NOESY (b)")
20
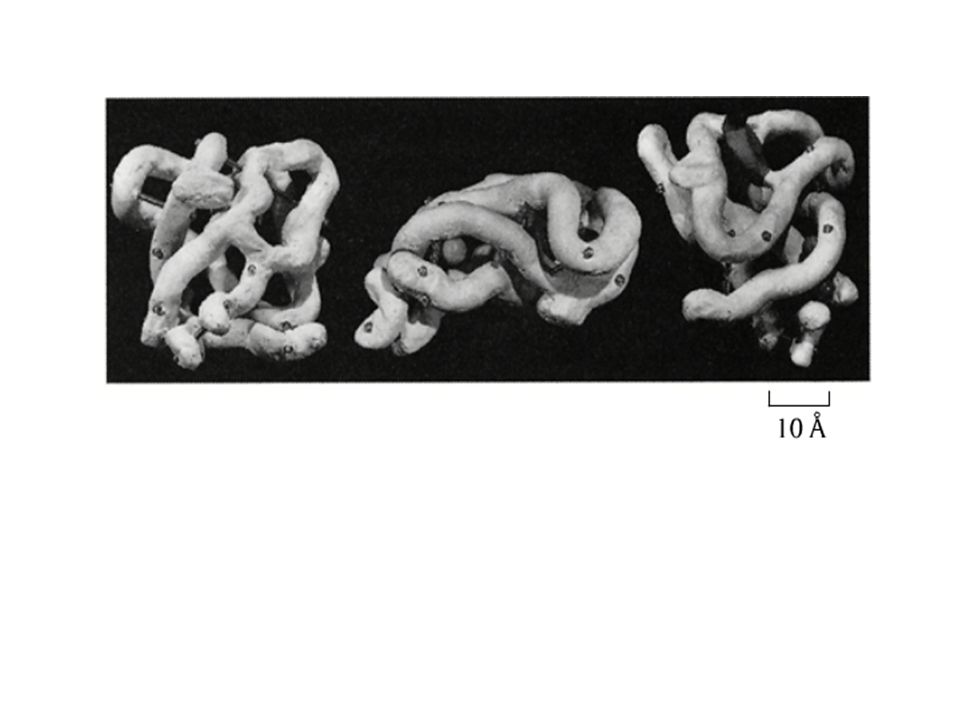
The information present in the NMR spectra can be used to calculate a 3D structure, which by virtue of the nature of the NMR data always is an ensemble of structures. The NMR data reveal more than X-ray data the flexibility present in protein structures. See the disorder in a few loops in the structure. See also the next slide.

24
The standard dimensions (Angstroms, degrees) of a planar trans-peptide group. The NH-CO groups form the peptide plane.

25
The Cis-peptide group.

26
A polypeptide chain in its fully extended form showing the planarity of the peptide groups

28
The torsional degrees of freedom of a peptide unit. The only resonable movements are around the Ca-N ( ) bond and the Ca-C bond
 bond and the Ca-C bond .")
29
Newman projections

31
Steric interactions between CO and the amide Hydrogen (NH) on adjacent residues prevents the occurrence of the conformation with = -60, = +30
 on adjacent residues prevents the occurrence of the conformation with = -60, = +30")
32
The Ramachandran diagram.

33
The conformation angle distribution of all residues (except Gly and Pro) in 12 very well determined X-ray structures.
 in 12 very well determined X-ray structures.")
34
A Ramachandran diagram of Gly residues. Note the large degree of freedom.

35
A right handed helix. The values are - 57 and = -47, 3.6 residues per turn and a pitch (distance between two points after complete turn) of 5.4 Angstrom. Hydrogen bonds between CO en NH are indicated by dashed lines. These hydrogen bonds are between residues N and N+4. This results in a strong hydrogen bond in a near optimal N....O distance of 2.8 Angstrom. The core of the helix is tightly packed, the sidechain groups all direct outward and avoid steric interference with the peptide backbone and with each other (next slide). A left-handed helix has a major problem: the side chains contact the backbone too closely. However, the Van der Waals overlap is not severe enough to avoid that 1-2 % of all non-Gly residues adopt this conformation.
 of 5.4 Angstrom. Hydrogen bonds between CO en NH are indicated by dashed lines. These hydrogen bonds are between residues N and N+4. This results in a strong hydrogen bond in a near optimal N....O distance of 2.8 Angstrom. The core of the helix is tightly packed, the sidechain groups all direct outward and avoid steric interference with the peptide backbone and with each other (next slide). A left-handed helix has a major problem: the side chains contact the backbone too closely. However, the Van der Waals overlap is not severe enough to avoid that 1-2 % of all non-Gly residues adopt this conformation..")
41
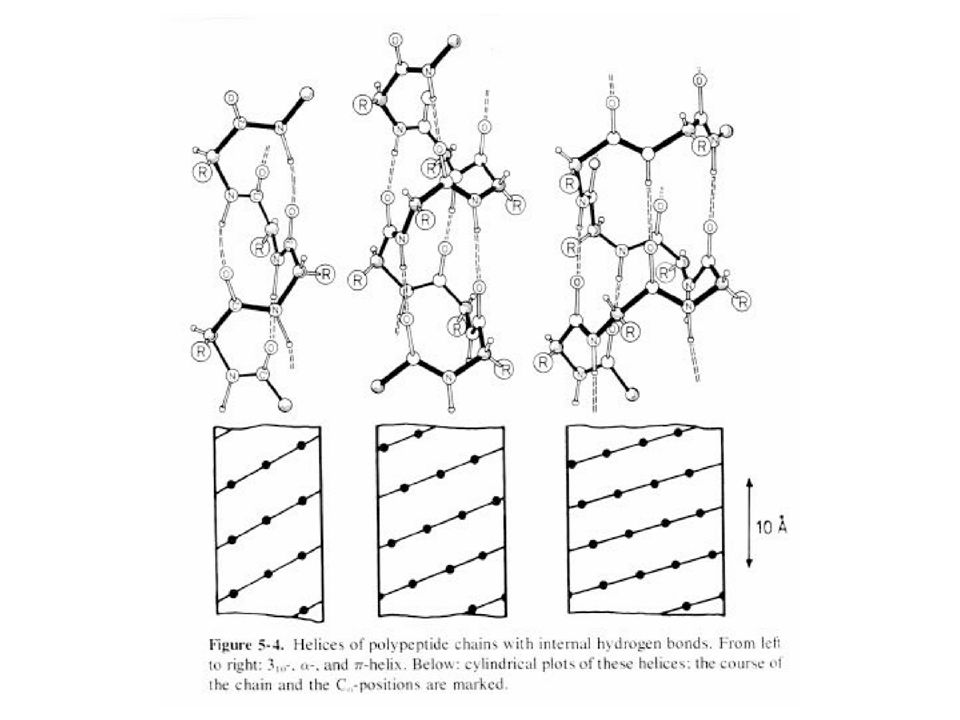
The hydrogen bond pattern of several polypeptide helices.

42
The 3-10 helix has 3.0 peptide units per turn with a pitch of 6.0 Angstrom. The helix has 3.6 units per turn and a pitch of 5.4 Angstrom The helix has 4.4 residues per turn and a pitch of 5.2 Angstrom Alle helices can be observed in proteins.

45
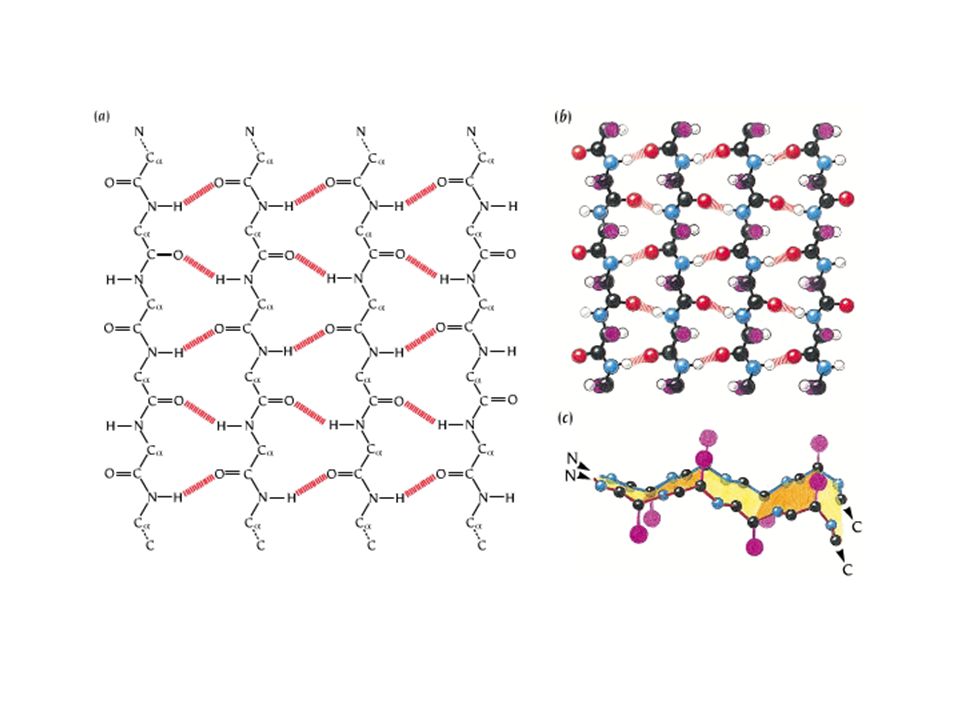
The hydrogen bond associations in pleated sheets.

46
A two-stranded -antiparellel pleated sheet. Dashed lines indicate hydrogen bonds. Note that the R groups (purple) on each polypeptide chain altenatively extend to opposite sides of the sheet.
 on each polypeptide chain altenatively extend to opposite sides of the sheet..")
48
Schematic representation of silk -sheets.

Similar presentations


















>")

















