Download presentation
Presentation is loading. Please wait.

1
Endomembrane System Protein Sorting and Transportation Shantou University Medical College Liu Gefei ( 刘戈飞 )
")
2
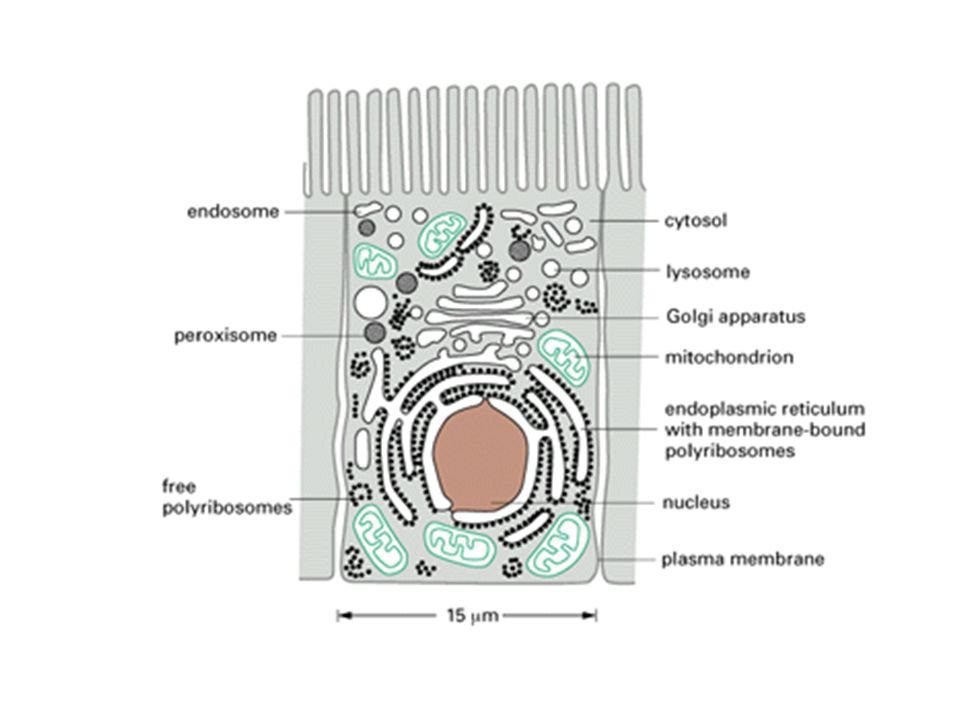
All living organisms are made up of cells. The eukaryotic cell contains a number of different types of organelles each of which is surrounded by a tightly sealed memberane.

3
The organization of a cell can be compared to that of a big city. In order to reach its correct destination, a letter has to be provided with an address label and a zip code, similar to the address tags on proteins.

5
Protein Transport Transmembrane transport Vesicular transport Gated transport cotranslational transloction endoplasmic reticlum (ER) post-translational mitochondira, peroxisome, ER Golgi complex, lysosome, cell membrane nucleus
 post-translational mitochondira, peroxisome, ER Golgi complex, lysosome, cell membrane nucleus")
6
Cell Traffic

7
Endoplastic Reticulum Transmemebrane transport

8
rough endoplasmic reticulum, rER the start site of secretory protein transport smooth endoplasmic reticulum, sER the calcium pool lipid synthesis

9
Translocation of secretory protein across ER membrane – signal hypothesis and sgnal peptide – process – major topological classes of integral membrane proteins Chaperone and chaperonin—protein folding and degradation –unfolded protein response –ubiquitin/proteasome pathway –amyloid plaque Amphipathic N-terminal signal sequences direct proteins to the mitochondrial matrix Sorting of peroxisomal proteins Targeting sequences

10
Translocation of secretory protein across ER membrane

11
Günter Blobel, born in 1936, works at the Laboratory of Cell Biology, The Rockefeller University, New York

12
1999 Nobel Physiology or Medicine Laureate Günter Blobel receiving his Nobel Prize from His Majesty the King Günter Blobel – Prize Award Photo Günter Blobel - Nobel Diploma

13
Press Release: The 1999 Nobel Prize in Physiology or Medicine NOBELFÖRSAMLINGEN KAROLINSKA INSTITUTET THE NOBEL ASSEMBLY AT THE KAROLINSKA INSTITUTE 11 October 1999 The Nobel Assembly at Karolinska Institutet has today decided to award the Nobel Prize in Physiology or Medicine for 1999 to Günter Blobel for the discovery that "proteins have intrinsic signals that govern their transport and localization in the cell" "The signal hypothesis"

14
In many inherited diseases, proteins are mislocalized in the cell due to errors in targeting signals and transport. One example is "primary hyperoxaluria," a rare disease, which results in kidney stones already at an early age. A signal in the enzyme alanine:glyoxylate aminotransferase normally directs it to the peroxisome. In patients, this signal is altered and the protein is mislocalized to the mitochondrion where it is unable to perform its normal function.

15
Today many important protein drugs (e.g. growth hormone, erythropoetin, insulin) are produced in living cells. To facilitate easy purification, the proteins are provided with a signal peptide causing them to be secreted out of the cell. For scale-up production, cells are grown in bioreactors.
 are produced in living cells. To facilitate easy purification, the proteins are provided with a signal peptide causing them to be secreted out of the cell. For scale-up production, cells are grown in bioreactors..")
16
Synthesis of secreted proteins (enzymes destined for the ER, Golgi complex, or lysosome and integral plasmamembrane proteins) begins on cytosolic ribosomes, which become attached to the membrane of the ER, forming the rough ER. The ER signal sequence on a nascent secretory protein consists of a segment of hydrophobic amino acids, generally located at the N-terminus.

17
In cotranslational translocation, the signal- recognition particle (SRP) first recognizes and binds the ER signal sequence on a nascent secretory protein and in turn is bound by an SRP receptor on the ER membrane, thereby targeting the ribosome/nascent chain complex to the ER. The SRP and SRP receptor then mediate insertion of the nascent secretory protein into the translocon. Hydrolysis of GTP by the SRP and its receptor drive this docking process.

18
As the ribosome attached to the translocon continues translation, the unfolded protein chain is extruded into the ER lumen. No additional energy is required for translocation.

22
In post-translational translocation, a completed secretory protein is targeted to the ER membrane by interaction of the signal sequence with the translocon. The polypeptide chain is then pulled into the ER by a ratcheting mechanism that requires ATP hydrolysis by the chaperone BiP, which stabilizes the entering polypeptide.

24
In both cotranslational and post-translational translocation, a signal peptidase in the ER membrane cleaves the ER signal sequence from a secretory protein soon after the N- terminus enters the lumen.

25
Major topological classes of integral membrane proteins synthesized on the rough ER

26
Synthesis and insertion into the ER membrane of type I single-pass proteins

27
Synthesis and insertion into the ER membrane of type II single-pass proteins.

28
Arrangement of topogenic sequences in single-pass and multipass membrane proteins inserted into the ER membrane The difference in the orientation of these proteins depends largely on whether there is a high density of positively charged amino acids(+++) on the N-terminal side of the SA sequence (type II) or onthe C-terminal side of the SA sequence (type III)
 on the N-terminal side of the SA sequence (type II) or onthe C-terminal side of the SA sequence (type III)")
29
Chaperones and Chaperonins Chaperones, which bind and stabilize unfolded or partly folded proteins, thereby preventing these proteins from aggregating and being degraded Chaperonins, which directly facilitate the folding of proteins.

30
Both of the two molecules do not contain the folding information of their facilitated proteins and are absent in the folded or mature proteins.

32
Only properly folded proteins and assembled subunits are transported from the rough ER to the Golgi complex in vesicles. The accumulation of abnormally folded proteins and unassembled subunits in the ER can induce increased expression of ER protein-folding catalysts via the unfolded protein response.

33
The unfolded-protein response

34
Unassembled or misfolded proteins in the ER often are transported back through the translocon to the cytosol, where they are degraded in the ubiquitin/proteasome pathway. pathway

35
Alternatively Folded Proteins Are Implicated in slowly Developing Diseases As noted earlier, each protein species normally folds into a single, energetically favorable conformation that is specified by its amino acid sequence.

36
Recent evidence suggests, however, that a protein may fold into an alternative three- dimensional structure as the result of mutations, inappropriate post-translational modification, or other as-yet-unidentified reasons. Such “misfolding” not only leads to a loss of the normal function of the protein but also marks it for proteolytic degradation.

37
The subsequent accumulation of proteolytic fragments contributes to certain degenerative diseases characterized by the presence of insoluble protein plaques in various organs, including the liver and brain.

38
Some neurodegenerative diseases, including Alzheimer’s disease and Parkinson’s disease in humans and transmissible spongiform encephalopathy (“mad cow” disease) in cows and sheep, are marked by the formation of tangled filamentous plaques in a deteriorating brain.
 in cows and sheep, are marked by the formation of tangled filamentous plaques in a deteriorating brain.")
39
Amyloid plaque

40
Amphipathic N-Terminal Signal Sequences Direct Proteins to the Mitochondrial Matrix

41
Protein import into the mitochondrial matrix

43
Three pathways for transporting proteins from the cytosol to the inner mitochondrial membrane

44
Two pathways for transporting proteins from the cytosol to the mitochondrial intermembrane space

46
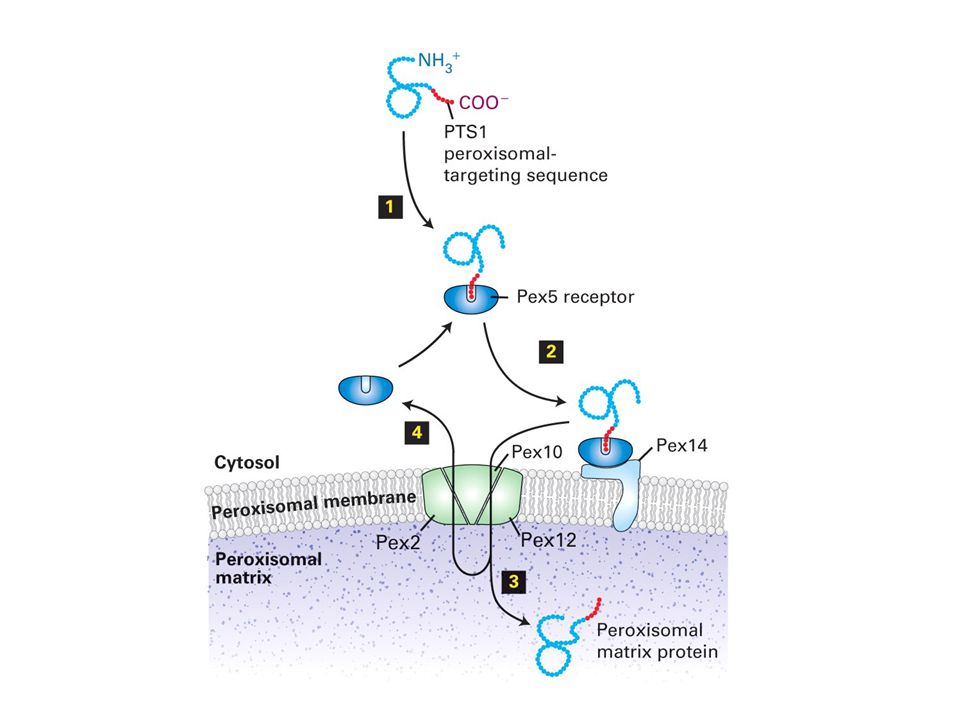
Sorting of Peroxisomal Proteins SKL sequence at the C-terminus of peroxisomal matrix proteins is known as peroxisomal-targeting sequence 1,simply PTS1. Most peroxisomal matrix proteins contain a C- terminal PTS1 targeting sequence; a few have an N-terminal PTS2targeting sequence. Neither targeting sequence is cleaved after import. Cytosolic receptor targets proteins with PTS into peroxisomal matrix.

48
Fluorescent-antibody staining of peroxisomal biogenesis mutants reveals different pathways for incorporation of membrane and matrix proteins

49
All proteins destined for the peroxisomal matrix bind to a cytosolic receptor, which differs for PTS1- and PTS2-bearing proteins, and then are directed to common import receptor and translocation machinery on the peroxisomal membrane.

50
Translocation of matrix proteins across the peroxisomal membrane depends on ATP hydrolysis. Many peroxisomal matrix proteins fold in the cytosol and traverse the membrane in a folded conformation.

51
Proteins destined for the peroxisomal membrane contain different targeting sequences than peroxisomal matrix proteins and are imported by a different pathway. Unlike mitochondria and chloroplasts, peroxisomes can arise de novo from precursor membranes, as well as by division of preexisting organelles.

54
Golgi Complex Vesicular transport

56
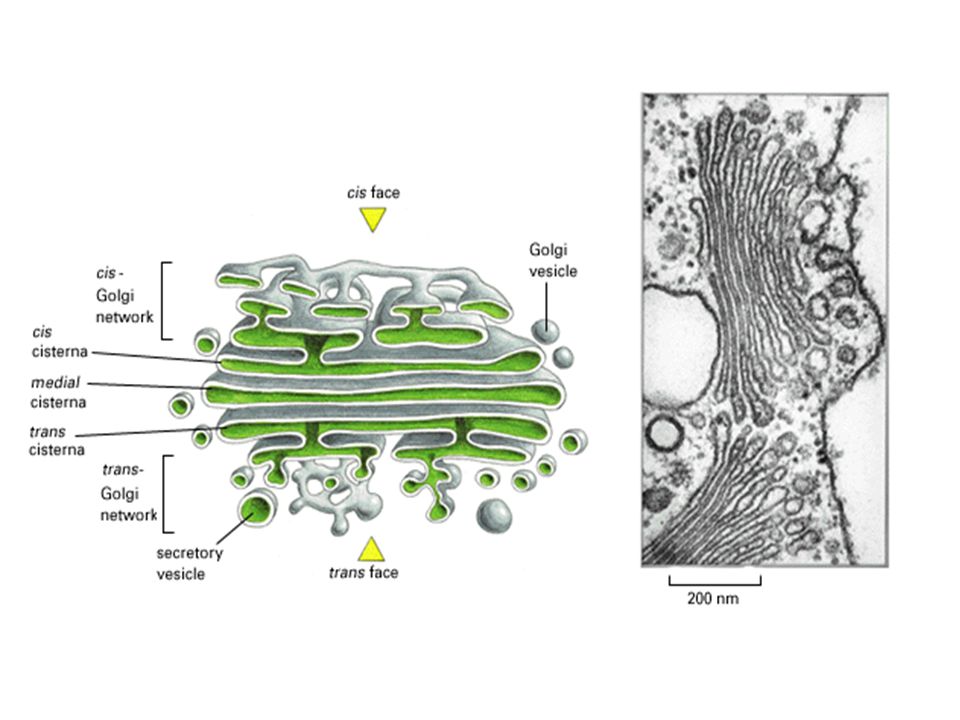
A polarized organelle –the transport direction in Golgi cis vesicules to cis to cisterna to trans to trans vesicules –specialized compartments cis network: protein sorting and O-link glycosylation media cisterna: protein modificationg and synthesize polysaccharides, lipids trans network: protein sorting and modification

57
Many Proteins Undergo Chemical Modification of Amino Acid Residues Acetylation phosphorylation glycosylation hydroxylation γ-carboxylation methylation

58
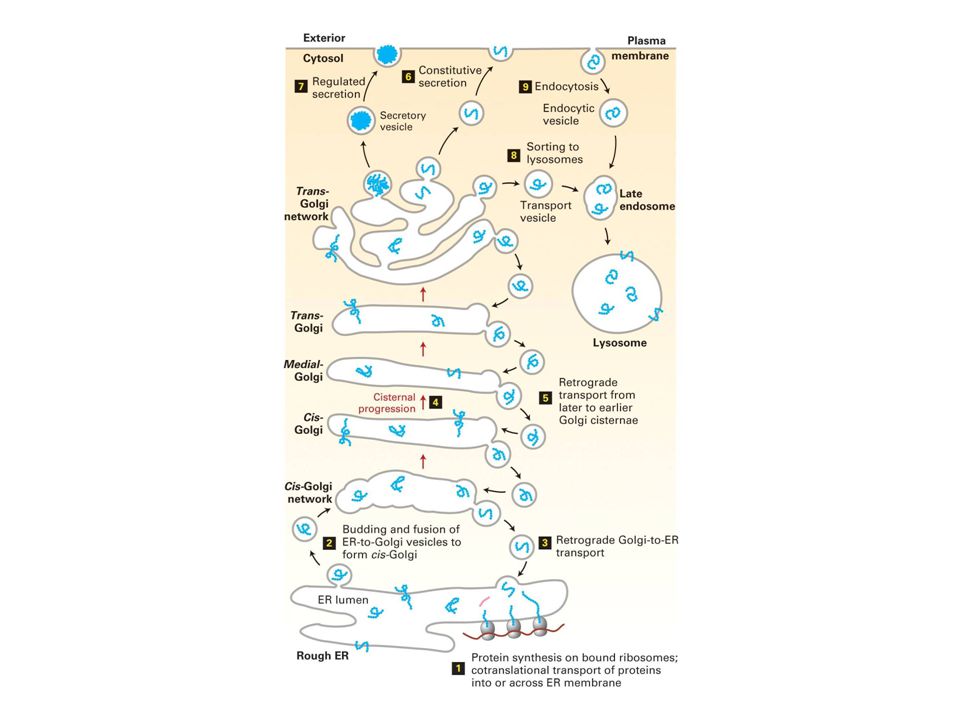
Vesicular transport Golgi complex is the center for protein sorting Clathrin vesicules transport protein from Golgi to lysosome Receptor mediated endocytosis vesicules involved in protein trafficing

59
Golgi complex- the center for protein sorting

61
Clathrin vesicules transport protein from Golgi to lysosome

62
Clathrin

63
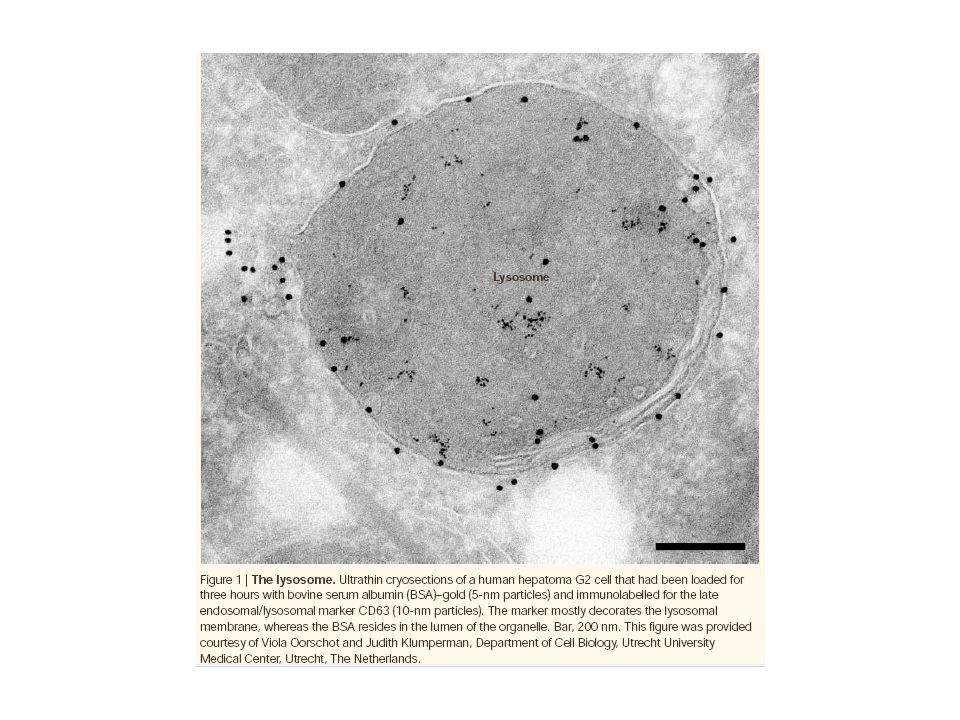
lysosomes membrane-limited organelles whose acidic interior is filled with hydrolytic enzymes. Lysosomal degradation is directed primarily toward extracellular proteins taken up by the cell and aged or defective organelles of the cell.

64
lysosome

65
acidmatrix

66
pH sensitive probe

67
1. 溶酶体的类型

69
吞噬

70
自噬

73
Receptor mediated endocytosis

75
vesicules involved in protein traffic

Similar presentations
















 mechanisms.>")








 Post -translational: proteins of plastids, mitochondria,>")




 bring amino acids (AA),>")

