Download presentation
Presentation is loading. Please wait.

1
Myotonic dystrophy DM Suhail Abdulla AlRukn

2
Outline Overview of repeat expansion disorders. Introduction to DM
Etiology and pathophysiology of DM Model mechanisms for myotonic dystrophy: Haploinsufficiency of DMPK Haploinsufficiency of SIX5 and neighboring genes RNA pathogenesis

3
Learning Objective Introduction to repeat expansion disorders and the different between the unstable expanding repeats in noncoding regions and ustable expanding repeats in coding regions. The pathophysiology of DM, and the three model mechanisms for myotonic dystrophy.

4
Overview of repeat expansion disorders
There are more than 30 neurological disorders that can attribute their pathogenesis to extensions of tandem repeats above a critical size. Yet the theories as to how large repeat arrays can cause such highly variable diseases are unresolved.

5
Overview of repeat expansion disorders
Most of the expansion disorders exhibit a delayed onset form of their diseases, indicating that they may share mechanisms that postpone clinical expression until later in life. Depending on where the unstable repeats are located within the gene, the repeat expansion disorders can be classified as having coding or noncoding mutations.

6
Overview of repeat expansion disorders
Coincidentally, in 1991, the first two triplet repeat expansion disorders discovered revealed examples from both coding and noncoding categories: The fragile X syndrome was linked to unstable CGG repeats in the noncoding 5′UTR of FMR1. Whereas spinobulbar muscular atrophy was associated with unstable coding CAG repeats.

7
Overview of repeat expansion disorders
- DM1 followed in 1992 as the third trinucleotide repeat expansion disorder discovered and was mapped to CTG repeats in the noncoding 3′UTR of DMPK. UREDs can be divided into two major classes: Unstable expanding repeats in noncoding regions. Unstable expanding repeats in coding regions.

8
Unstable expanding repeats in noncoding regions.
Biochim Biophys Acta Feb;1772(2): Epub 2006 Jun 20.
: Epub 2006 Jun 20.")
9
Unstable expanding repeats in coding regions.
Biochim Biophys Acta Feb;1772(2): Epub 2006 Jun 20.
: Epub 2006 Jun 20.")
10
Introduction Myotonic dystrophy (DM) is the most common muscular dystrophy in adults, and is the second most common muscular dystrophy after Duchenne muscular dystrophy. Onset is usually in the second or third decade. The prevalence of DM is 1 in 8000 in the general population
 is the most common muscular dystrophy in adults, and is the second most common muscular dystrophy after Duchenne muscular dystrophy. Onset is usually in the second or third decade. The prevalence of DM is 1 in 8000 in the general population.")
11
The clinical signs of classical myotonic dystrophy
Netter, F.H. The CIBA collection of Medical Illustrations. 1986

12
Types The genetic causes of three forms of DM have been identified:
DM1, also known as Steinert's disease DM2, also known as proximal myotonic myopathy (PROMM) 1994 Congenital myotonic dystrophy (CMyD)
 Congenital myotonic dystrophy (CMyD)")
13
POPULATION GENETICS In the Saguenay region of the province of Quebec, the prevalence of DM is about 1 in 475; about 600 cases are known in a population of 285,000. The prevalence of myotonic dystrophy is 30 to 60 times higher than the prevalence in most other regions of the world. (1/475 Vs 1/25,00).
.")
14
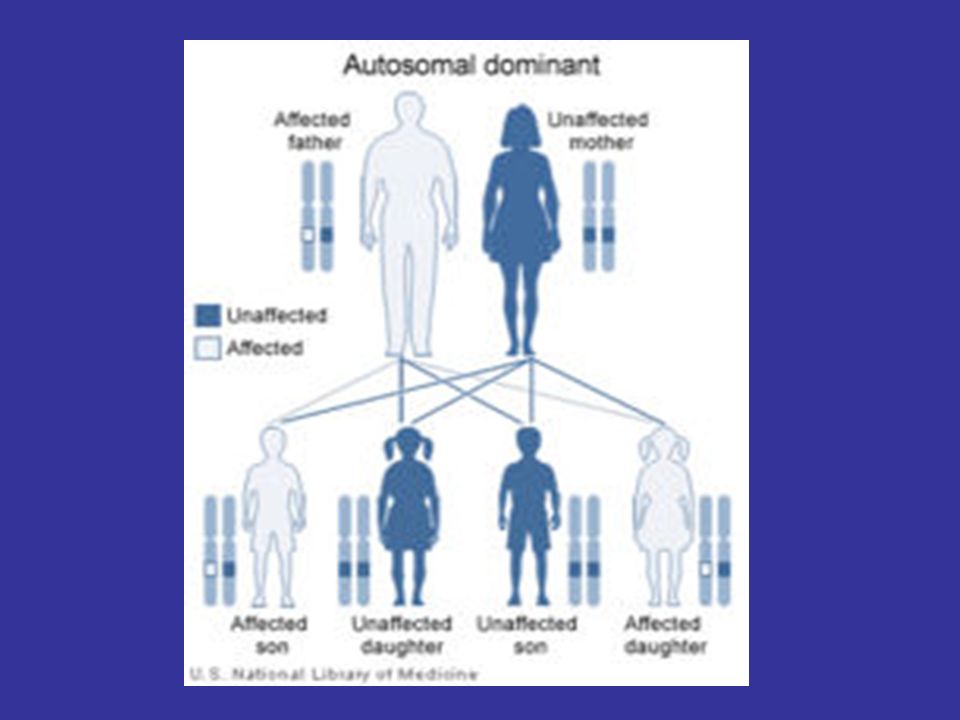
Etiology DM is autosomal dominant disease.
DM1 results from an unstable trinucleotide repeat expansion, CTG, in the 3' non-coding region (myotonic dystrophy type-1 protein kinase, DMPK) on chromosome 19q13.3. - DM2 results from an unstable tetranucleotide (CCTG) repeat expansion in the intron 1 of ZNF9 on chromosome 3q21.
 on chromosome 19q DM2 results from an unstable tetranucleotide (CCTG) repeat expansion in the intron 1 of ZNF9 on chromosome 3q21.")
16
Etiology Source: Science (2001) 293:
 293:")
17
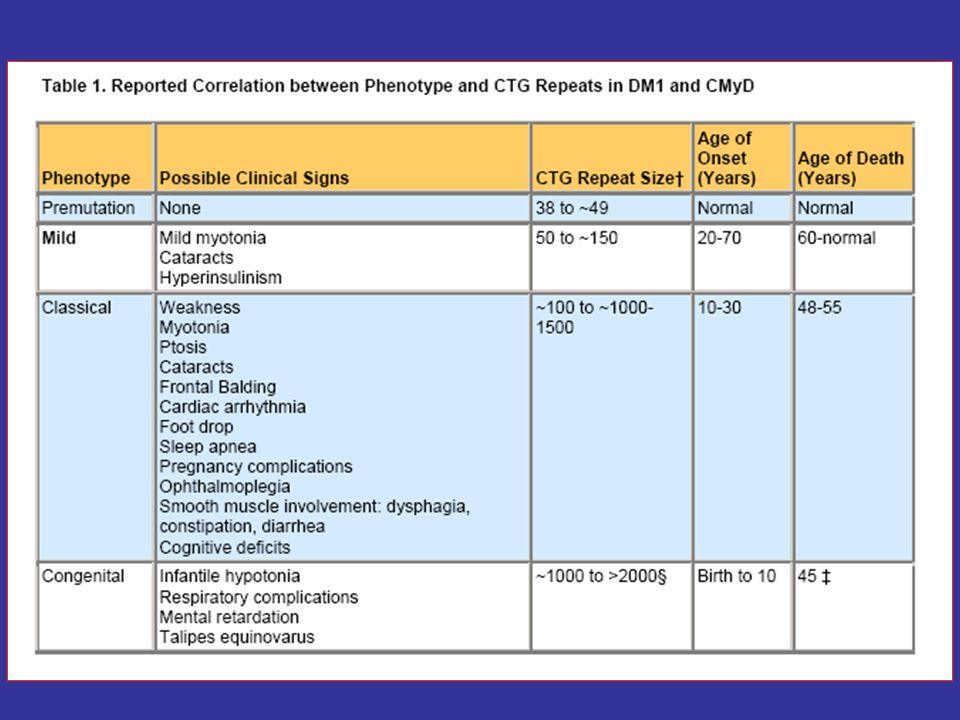
Etiology The cause of the unstable CTG repeat expansion is unknown; however, it is thought to occur during gametogenesis and is more extensive when coming from a female carrier. - The disease severity generally correlates with repeat length.

18
Source: Science (2001) 293:
 293:")
20
Model mechanisms for myotonic dystrophy
Haploinsufficiency of DMPK Haploinsufficiency of SIX5 and neighboring genes RNA pathogenesis

21
RNA toxic gain-of-function model for myotonic dystrophy
Biochim Biophys Acta Feb;1772(2): Epub 2006 Jun 20.
: Epub 2006 Jun 20.")
22
Haploinsufficiency of DMPK
Early expression studies were consistent with the hypothesis that the mutation interfered with DMPK production, in that mRNA and protein levels were reduced in patient muscle and cell culture. However, DMPK knockout mice showed only a very mild, late-onset myopathy without the multisystemic features of the disease.

23
Haploinsufficiency of DMPK
The fact that no DMPK point mutations have been associated with a DM phenotype further suggests that the multisystemic features of DM1 are not simply caused by DMPK haploinsufficiency.

24
Haploinsufficiency of SIX5 and neighboring genes
A second proposed mechanism has been that the mutation interferes with expression of multiple genes in the DM1 region, possibly through regional effects produced by repeat-induced alterations in chromatin structure. In addition to DMPK and the neighboring homeodomain gene SIX5, other regional genes suggested to be involved in DM1 pathogenesis have included myotonic dystrophy gene with WD repeats, DMWD, which is prominently expressed in the testis and brain.

25
Haploinsufficiency of SIX5 and neighboring genes
In this model, the multisystemic features of DM1 would be explained by haploinsufficiency of a number of neighboring genes, with expression level and hence disease severity, dependent on repeat length. In support of this possibility, Six5 knockout mice develop cataracts, but without the posterior subcapsular location or the distinctive opacities that are characteristic of cataracts in DM patients.

26
RNA pathogenesis A third hypothesized mechanism is that the enlarged CUG-containing transcripts accumulate as intranuclear foci and disrupt cellular function. Direct support for this model came from a transgenic mouse model, in which the CTG expansion was inserted into the 30 end of the human skeletal actin gene, a gene not directly involved in DM1 but which is expressed only in skeletal muscle.

27
RNA pathogenesis This mouse model expressed an mRNA with a CUG repeat tract of 250 repeats, and caused the myotonia and myopathic features characteristic of DM1. But because CUG-containing transgene expression was limited to skeletal muscle the role of the CUG expansion in the multisystemic features of DM was not addressed.

28
RNA toxic gain-of-function model for myotonic dystrophy
Biochim Biophys Acta Feb;1772(2): Epub 2006 Jun 20.
: Epub 2006 Jun 20.")
29
Pathogenic model of DM1 and DM2.

30
Muscle histology in DM2

31
Home Take massage - The severity of the URED’s correlates with repeat length DM1 results from an unstable trinucleotide repeat expansion, CTG, DMPK on chromosome 19q13.3. DM2 results from an unstable tetranucleotide (CCTG) repeat expansion in the intron 1 of ZNF9 on chromosome 3q21. The cause of the unstable CTG repeat expansion is unclear but there are few model mechanisms.
 repeat expansion in the intron 1 of ZNF9 on chromosome 3q21. The cause of the unstable CTG repeat expansion is unclear but there are few model mechanisms.")
32
Thanks

Similar presentations




. Mary Beth Busby founding board member of the Fragile X Research Foundation (FRAXA) Walter Kaufmann Director,>")





 Elaine Lyon, Ph.D. University of Utah/ARUP Laboratories Association.>")

 Peters et al.>")






