Download presentation
Presentation is loading. Please wait.

1
Disorders of myelination and Neuronal storage disease
Leukodystrophies Demyelinating disease Metabolic neuronal storage disease

2
Diseases of myelin can be divided into two broad groups:
Dysmyelinating diseases Demyelinating diseases

3
Dysmyelinating Diseases
These disorders are also termed leukodystrophies, and almost all of them manifest themselves early in life and are genetically determined. Profound disturbance in the formation and preservation of myelin so that its proper functioning is never established.

4
Demyelinating Diseases
The myelin sheath, once properly formed and functioned, is destroyed by a disease process. The most common disease in this category is multiple sclerosis. Other examples include: Central pontine myelinolysis Progressive multifocal leukoencephalopathy Subacute combined degeneration of the spinal cord

5
Diseases of myelin Leukodystrophies (congenital)
Metachromatic Leukodystrophy Krabbe’s disease Adreno-Leukodystrophy (ALD-Lorenzo) Alexander disease Multiple sclerosis (acquired)
 Alexander disease. Multiple sclerosis (acquired)")
6
Leukodystrophies Similar to neuronal storage diseases: Most are lysosomal storage diseases with specific enzymatic defects (metachromatic leukodystrophy, Krabbe leukodystrophy) Different from neuronal storage diseases: White matter involvement Storage material is toxic : globoid cells (Krabbe disease), accumulates in macrophages (Metachromatic leukodystrophy).
 Different from neuronal storage diseases: White matter involvement. Storage material is toxic : globoid cells (Krabbe disease), accumulates in macrophages (Metachromatic leukodystrophy).")
7
Hallmarks: Lysosomal abnormalities--diagnosis based on enzyme defect, frequently recessive. White matter involvement--storage is not usually neuronal, symptoms relate to white matter involvement.

8
Clinical findings in Leukodystrophies
Similar to neuronal storage diseases The enzyme deficiency in Adrenoleukodystrophy, Metachromatic leukodystrophy and Krabbe Differences: Signs and symptoms relate to white matter abnormalities (pyramidal signs) Gait (walking) disorders Loss of motor abilities, Spasticity. Peripheral nerve involvement occurs in ALD, MLD and Krabbe’s disease.
 Gait (walking) disorders. Loss of motor abilities, Spasticity. Peripheral nerve involvement occurs in ALD, MLD and Krabbe’s disease.")
9
Metabolic Disorder Inheritance Abnormality Metachromatic leukodystrophy Autosomal Recessive Arylsulfatase A deficiency Krabbe disease Galactocerebroside β-galactosidase deficiency Adrenoleukodystrophy Autosomal Recessive, X-linked Peroxisomal defects; elevated very long chain fatty acids Canavan disease Aspartoacylase deficieny Pelizaeus-Merzbacher disease X-linked Mutations in proteolipid protein Vanishing white matter disease Translation initiation factor; link to myelin unclear Alexander disease Mutations in glial fibrillary acidic protein

10
Metachromatic Leukodystrophy
Autosomal recessive Presents in infancy, Most common of the leukodystrophies Both central and peripheral white matter involved Course is progressive, usually fatal in a few years Pathology is diffuse, confluent loss of myelin that is most advanced in the cerebrum.

11
Due to inborn error of metabolism in which arylsulfatase A, although present within lysosomes, is enzymatically inactive. Leads to breakdown of myelin and the accumulation of galactosyl sulfatides (cerebroside) within schwann cells and oligodendrocytes The metachromatic material (sulfatide) stains brown with toluidine blue, cresyl violet, thionin or acriflavine (blue stains).
 within schwann cells and oligodendrocytes. The metachromatic material (sulfatide) stains brown with toluidine blue, cresyl violet, thionin or acriflavine (blue stains).")
12
The metachromatic material (sulfatide) stains brown with toluidine blue, cresyl violet, thionin or acriflavine (blue stains).
 stains brown with toluidine blue, cresyl violet, thionin or acriflavine (blue stains).")
13
Krabbe Disease Globoid cell leukodystrophy
Usually appears in early months of life and progresses to death in one to two years. Motor signs (hypertonic flexion), optic atrophy. Autosomal recessive Caused by deficiency of b-galactosidase. Galactocerebroside accumulates and expressed histologically by the presence of perivascular aggregates of globoid cells: Undigested galactocerebroside in globoid cells (macrophages) Loss of oligodendrocytes.
, optic atrophy. Autosomal recessive. Caused by deficiency of b-galactosidase. Galactocerebroside accumulates and expressed histologically by the presence of perivascular aggregates of globoid cells: Undigested galactocerebroside in globoid cells (macrophages) Loss of oligodendrocytes.")
14
Adreno-Leukodystrophy (ALD)
Severe, bilateral, symmetric loss of myelin Aut. Rec. & X-linked Presents in childhood (3-10 years), lethal in a few years High levels of very long chain fatty acids Adrenoleukodystrophy is peroxisomal*. *The peroxisome is a cellular organelle measuring 0.5 micron in diameter that participates in important cellular functions such as beta-oxidation of very-long-chain fatty acids (VLCFA), plasmalogen production, and bile acid synthesis.
, lethal in a few years. High levels of very long chain fatty acids. Adrenoleukodystrophy is peroxisomal*. *The peroxisome is a cellular organelle measuring 0.5 micron in diameter that participates in important cellular functions such as beta-oxidation of very-long-chain fatty acids (VLCFA), plasmalogen production, and bile acid synthesis.")
15
Adrenoleukodystrophy (ALD)
Adrenal insufficiency pigment, diarrhea, hypotension Peroxisomal membrane disorder High levels of very long chain fatty acids in tissue and fluids Lorenzo’s oil contains short chain FA’s VLCFA also seen in schwann cells and macrophages in the demyelinated CNS.

17
Alexander Disease Loss of myelin with numerous Rosenthal fibers
refractile eosinophilic hyaline bodies found within the cytoplasm of astrocytes particularly in the subpial, subependymal, and perivascular regions. Myelin is preserved in peripheral nervous system Periventricular white matter of frontal lobes Loss of myelin

19
Demyelinating disease
Acquired not congenital Mechanism is autoimmune not metabolic Hallmark is the plaque of abrupt demyelination Common locations: optic nerves and chiasm and paraventricular white matter

20
Demyelinating diseases
Multiple sclerosis MS variants: DeVic, Marburg Acute disseminated encephalomyelitis (acute) Acute necrotizing hemorrhagic encephalomyelitis (hyper-acute) Central pontine myelinolysis Marchiafava-Bignami
 Acute necrotizing hemorrhagic encephalomyelitis (hyper-acute) Central pontine myelinolysis. Marchiafava-Bignami.")
21
Multiple Sclerosis First “attack” may be a single symptom, commonly optic neuritis: Ophthalmoplegia Monocular blindness. Facial hypesthesia or trigeminal neuralgia (tic douloureux) Bell’s palsy, hemifacial spasm, vertigo, vomiting, nystagmus, deafness, abnormal speech, intention tremor, ataxia, motor abnormalities, bowel and bladder dysfunction.
 Bell’s palsy, hemifacial spasm, vertigo, vomiting, nystagmus, deafness, abnormal speech, intention tremor, ataxia, motor abnormalities, bowel and bladder dysfunction.")
22
Hallmark: demyelinating plaques, peripheral nerves spared.
Multiple sclerosis Temperate climates (rare in tropics with increasing frequency further from equator) F>M (x2), mean age = 30 Pathogenesis: genetic predisposition (HLA-DR2), auto- immune, viral—EPV Hallmark: demyelinating plaques, peripheral nerves spared.
 F>M (x2), mean age = 30. Pathogenesis: genetic predisposition (HLA-DR2), auto- immune, viral—EPV. Hallmark: demyelinating plaques, peripheral nerves spared.")
23
Pathology of MS Multiple plaques
These are sharply delineated, irregular zones of total demyelination with initial preservation of axons. They are most numerous in the white matter of the cerebrum (periventricular), brain stem, cerebellum and spinal cord (peripheral regions). Within the plaque, initially axons are preserved. Microglial cells proliferate and phagocytize the myelin debris. Later, a glial scar forms.
, brain stem, cerebellum and spinal cord (peripheral regions). Within the plaque, initially axons are preserved. Microglial cells proliferate and phagocytize the myelin debris. Later, a glial scar forms.")
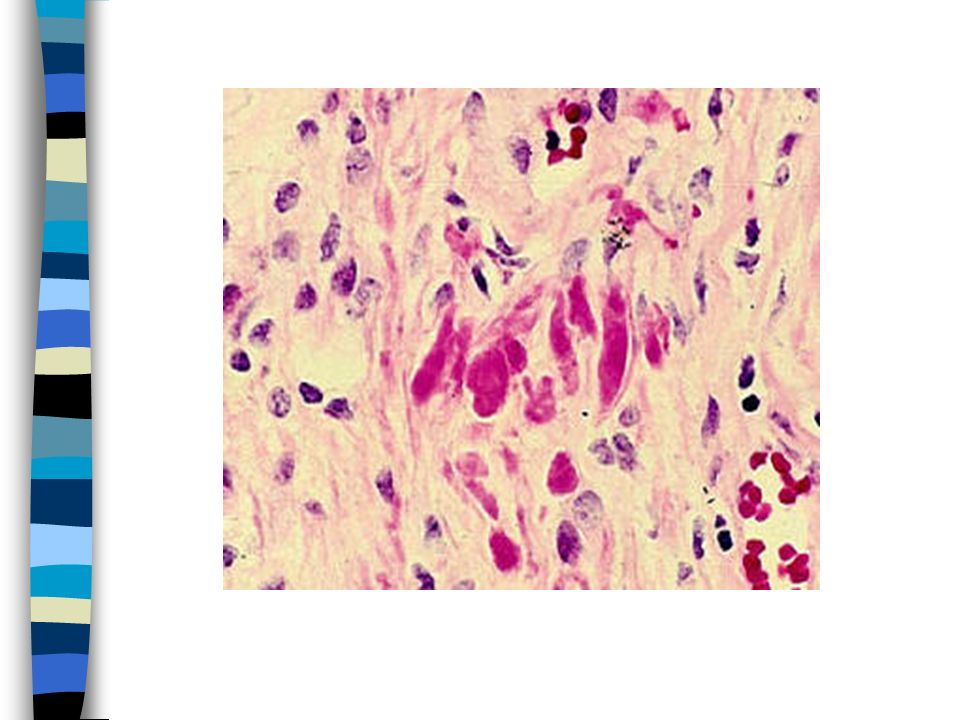
24
PLAQUES OF DEMYELINATION
MULTIPLE SCLEROSIS PLAQUES OF DEMYELINATION MYELIN STAIN

25
PLAQUES OF DEMYELINATION
MULTIPLE SCLEROSIS PLAQUES OF DEMYELINATION

26
Demyelinating diseases
Post-infectious encephalomyelitis perivascular demyelination with lymphocytes, ?auto-immune headache, vomiting fever, 15-20% die similar to “post-vaccinal encephalomyelitis”. Central pontine myelinolysis quadriparesis, pseudo-coma usually in alcoholics.

27
Summary of Primary Diseases of Myelin
Because of the critical role of myelin in nerve conduction, diseases of myelin can lead to widespread and severe neurologic deficits. Diseases of myelin can be grouped into demyelinating diseases (in which normal myelin is broken down for inappropriate reasons-often by inflammatory processes), and dysmyelinating diseases (which are metabolic disorders that include the leukodystrophies in which the underlying struc-ture of the myelin is abnormal or its turnover is abnormal). Multiple sclerosis, an autoimmune demyelinating disease, is the most common disorder of myelin, affecting young adults often with a relapsing-remitting course with eventual progressive accumulation of neurologic deficits. Other less common forms of immune-mediated demyelination often follow infections and are more acute illnesses
, and dysmyelinating diseases (which are metabolic disorders that include the leukodystrophies in which the underlying struc-ture of the myelin is abnormal or its turnover is abnormal). Multiple sclerosis, an autoimmune demyelinating disease, is the most common disorder of myelin, affecting young adults often with a relapsing-remitting course with eventual progressive accumulation of neurologic deficits. Other less common forms of immune-mediated demyelination often follow infections and are more acute illnesses.")
28
Neuronal storage diseases

29
Neuronal storage diseases
Storage material typically within neurons Psychomotor retardation Mental retardation Most are lysosomal Most are autosomal recessive Enzymatic deficiencies.

30
Neuronal storage disease
In general: Visual impairment (due to retinal pathology) Seizures are more common (vs. leukodystrophies) due to neuronal involvement Some show hepatosplenomegaly: Gaucher disease Hurler syndrome Niemann-Pick disease.
 Seizures are more common (vs. leukodystrophies) due to neuronal involvement. Some show hepatosplenomegaly: Gaucher disease. Hurler syndrome. Niemann-Pick disease.")
31
Tay-Sachs disease accumulation of ganglioside in lysosomes
Hexosaminidase A deficiency absent hexosaminidase A&B = Sandhoff’s disease. accumulation of ganglioside in lysosomes cherry red spot on retina myelin figures in lysosomes on EM (membranous cytoplasmic bodies).
.")
32
Hurler Syndrome Autosomal recessive
Neuronal accumulation of mucopolysaccharides Visceral organs can be involved hepatosplenomegaly Associated features include: dwarfism, corneal opacities skeletal deformities.

33
Gaucher disease Glucocerebroside in macrophages Autosomal recessive
Most common of the sphingolipidoses Adult.

34
Many inherited disorders of metabolism can lead to accumulation of storage products in cells, as seen here with Gaucher's disease involving spleen. The large pale cells contain an accumulated storage product from lack of an enzyme.

35
Niemann-Pick disease Sphingomyelinase deficiency causes sphingomyelin accumulation within mononuclear phagocyte system (and neurons and glial cells) Autosomal recessive cherry red spot similar to Tay-Sachs disease.

36
Leukodystrophy vs. Neuronal storage
Leukodystrophies: Adrenoleukodystrophy, Metachromatic leukodystrophy, Krabbe disease, Alexander disease white matter rarefaction, motor findings, enzymatic defect. Neuronal storage diseases: Gaucher disease, Nieman Pick, Tay Sachs, Hurler syndrome storage material in neurons, cherry red spot, seizures, enzymatic defect.

37
Thank you!

Similar presentations

 (MND) are a group of neurological disorders that selectively affect motor neurons.>")












 Name: Helen Weezy F Baby Age: 28 Symptoms: Strange pricks in her hands and feet, fatigue, impaired.>")
 involves an immune-mediated process in which an abnormal response of the body’s immune system.>")


 on nerve fibers in the brain is lost,>")

