Download presentation
Presentation is loading. Please wait.

1
Newborn Screening Historical, Ethical, Technological Aspects
Nutrition 526 November 9, 2009 Cristine M Trahms, MS, RD Beth Ogata, MS, RD Lisa Sniderman King, CGC

2
First screen must be taken 24-48 hours of life regardless of feeding status or weight
Blood Sample on Guthrie Filter Paper Card

3
Unsatisfactory Specimens
(Provided by the New York State Department of Health) Clotted or Layered Serum Rings Specimen Not Dried Before Mailing Supersaturated No Blood Diluted, Discolored, or Contaminated Scratched or Abraded Quantity Insufficient for Testing
 Clotted or Layered. Serum Rings. Specimen Not Dried Before Mailing. Supersaturated. No Blood. Diluted, Discolored, or Contaminated. Scratched or Abraded. Quantity Insufficient for Testing.")
4
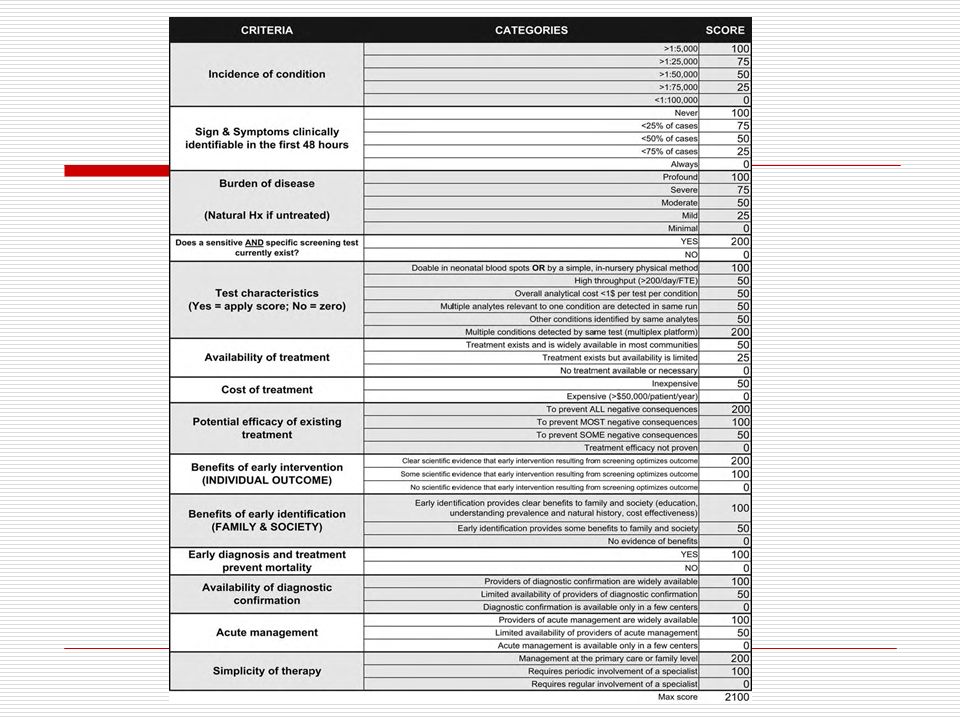
Criteria for Newborn Screening
important condition acceptable treatment available facilities for diagnosis and treatment difficult to recognize early suitable screening test natural history known cost effective to diagnose and treat Wilson & Jungner, 1968

5
MS/MS -High Impact and High Throughput
One disease, one test is not cost effective Many diseases, one test is cost effective MS/MS allows for rapid, simultaneous analysis and detection of many disorders of amino acid, organic acid, and fatty acid metabolism.

6
What is MS/MS ? Collision Cell Mass Spectrometer 2
3/16” blood spot deproteinization derivatization Mass Spectrometer 1 Mass Spectrometer 2 Collision Cell CH3 R - COO CH3 +N - CH2 - CH - CH2 - COO - C4H9 CH3 CH3 R - COO CH3 +N - CH2 - CH - CH2 - COO - C4H9 CH3 +CH2 - CH - CH2 - COOH (m/z 85) Data of all compounds within selected range ( m/z) Data of product ions with a mass of 85 only Data system correlates m/z 85 to its precursor ion’s mass and records the abundance of all precursors (parents of m/z 85)
 Data of all compounds. within selected range. ( m/z) Data of product ions. with a mass of 85 only. Data system correlates m/z 85 to its precursor ion’s mass and records the abundance of all precursors (parents of m/z 85)")
7
Tandem Mass Spectrometry (MS/MS)
Compounds analyzed are amino acids & acylcarnitines Amino acids – PKU, MSUD, Homocystinuria Acylcarnitine {Carnitine (vehicle) +fatty acid} for identification of organic acidurias and fatty acid oxidation disorders.
 +fatty acid} for identification of organic acidurias and fatty acid oxidation disorders.")
8
Amino Acid Disorders AA that are not used to make proteins are recycled by their specific metabolic pathways. Enzymatic deficiencies in these pathways lead to various clinical phenotypes. PKU – Phenylketonuria : severe perm MR MSUD – Maple syrup urine disease: dd, hallucinations, ataxia HCY – Homocystinuria: connective tissue damage – joints, heart, dd, psychiatric dist. CIT – Citrullinemia: risk of hyperammonemiadd, coma, death ASA – Argininosuccinic acidemia: brittle hair, liver dis, dd TYR I – Tyrosinemia type I: acute or chronic liver disease, liver cancer, neurologic pain crises. Diagnosed by plasma amino acids, and/or urine amino acids, and/or urine organic acids (takes 2-5 days)
")
9
Organic Acid Disorders
Organic acids are breakdown products of protein and fatty acid metabolism. Defects in their breakdown lead to (generally) Vomiting, metabolic acidosis, elevated ammonia in crises Dd, motor delay, ataxia, heart/kidney/pancreatic problems IVA - Isovaleric acidemia GA I – Glutaric acidemia type I HMG – 3-OH 3-CH3 glutaric aciduria MCD – Multiple carboxylase deficiency MUT – Methylmalonic acidemia (mutase def) 3MCC – 3-Methylcrotonyl-CoA carboxylase deficiency Cbl A,B – Methylmalonic acidemia PROP – Propionic acidemia BKT – Beta-ketothiolase deficiency Diagnosed by urine organic acids and/or plasma acylcarnitines.
 Vomiting, metabolic acidosis, elevated ammonia in crises. Dd, motor delay, ataxia, heart/kidney/pancreatic problems. IVA - Isovaleric acidemia. GA I – Glutaric acidemia type I. HMG – 3-OH 3-CH3 glutaric aciduria. MCD – Multiple carboxylase deficiency. MUT – Methylmalonic acidemia (mutase def) 3MCC – 3-Methylcrotonyl-CoA carboxylase deficiency. Cbl A,B – Methylmalonic acidemia. PROP – Propionic acidemia. BKT – Beta-ketothiolase deficiency. Diagnosed by urine organic acids and/or plasma acylcarnitines.")
10
Fatty Acid Oxidation Disorders
Fatty acid disorders lead to impaired energy production. Hypoglycemia, cardiomyopathy, muscle weakness can be seen MCAD – Medium-chain acyl-CoA dehydrogenase deficiency VLCAD – Very long-chain acyl-CoA dehydrogenase deficiency LCHAD – Long-chain L-3-OH acyl-CoA dehydrogenase deficiency TFP – Trifunctional protein deficiency CUD – Carnitine uptake defect Diagnosed by plasma acylcarnitines and urine organic acids can be helpful.

11
MS/MS Plasma Acylcarnitines
Intensity 100% * MCAD C2 C16 C8 C10:1 C6 * 100% Control C2 Intensity * internal standards

12
VLCAD profile C14:1 C12 C18:1 C16 Normal VLCAD * * * * * * * C16
Free Carnitine C2 * C12 * C8 * C4 * C3 * VLCAD Free Carnitine * C14:1 C12 C18:1 C16 * internal standards

13
MS/MS Plasma Amino Acids

14
Acylcarnitine – VLCAD Deficiency
Cutoff is 0.65

15
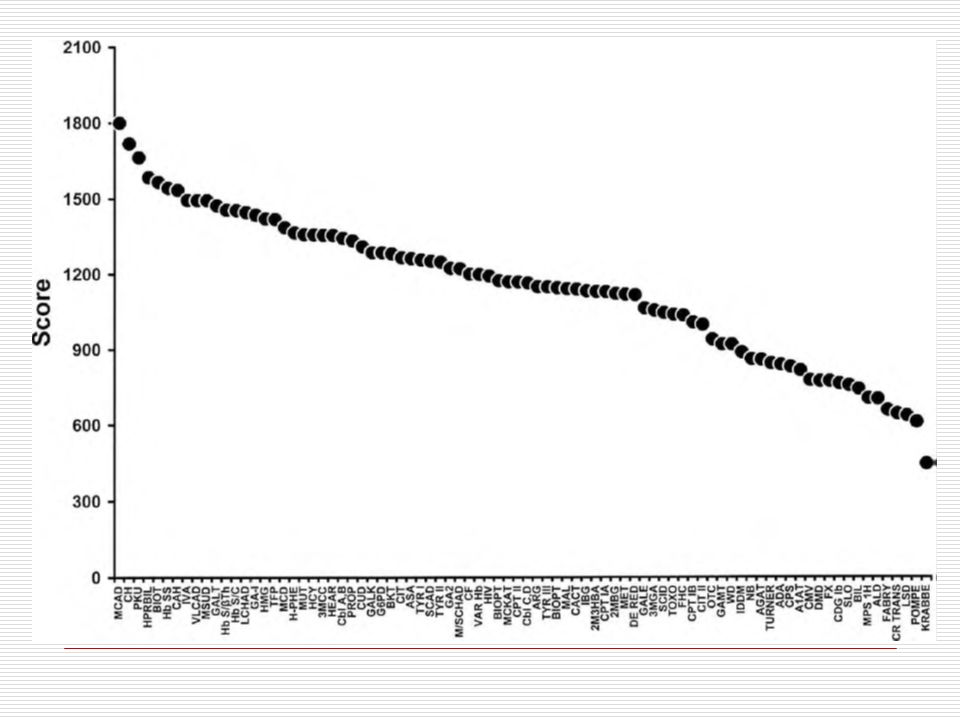
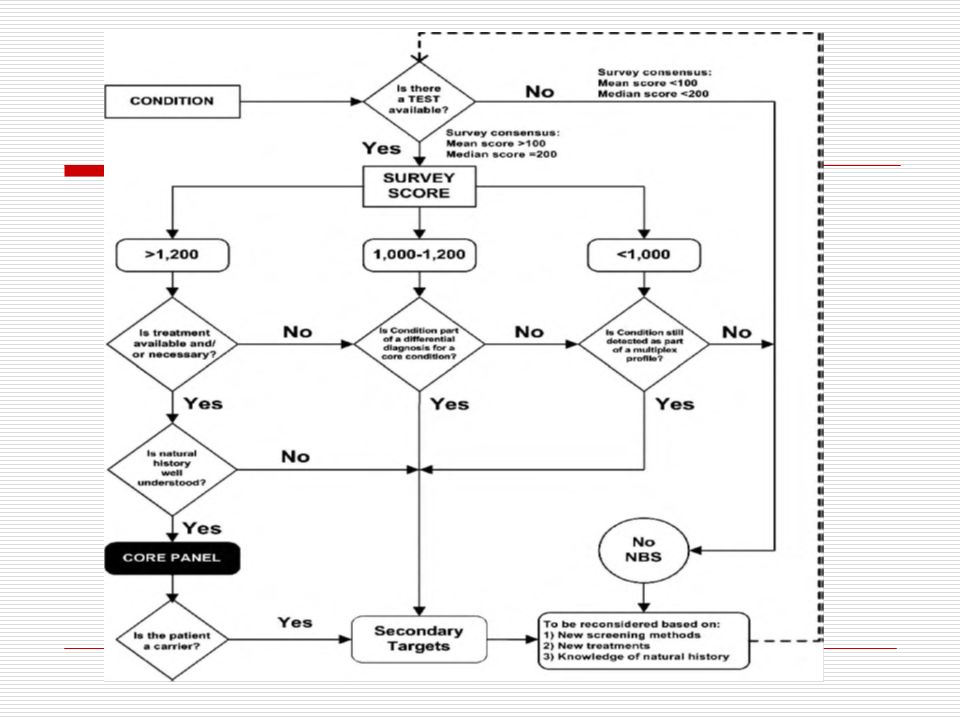
Which Disorders to Screen For?
NBS mandates are under state control Some states screened for 3 diseases, others 40+ 2002 Maternal and Child Health Bureau commissioned ACMG Analyze literature Develop consensus on which disorders Recommend a core panel to create uniform NBS across all states.

19
So why the need for intense deliberation, why not screen for everything?

20
Historical Harm (?) Early PKU screening led to cases of overly restricted phe and/or implementation of diet prior to confirmation of diagnosis Today, diagnosis is quite rapid 40 years ago it took much longer so more potential for harm However, no published evidence of wide-spread physical/medical harm BUT the cases do underscore need for expertise and resources for mgmt

21
Whom do we see? Patients who need active management
Symptomatic at diagnosis Strong evidence of pathology if untreated Examples: PKU, classic gal, MSUD, PA etc. Detecting these babies is the primary goal of the NBS program. A portion of these kids will die within the first week of life despite NBS if sample not collected promptly.

22
Whom do we see? Patients with disorders known to pose risk but reduced penetrance ie. probably not everyone needs to be treated HPHE, MCAD Both are/have mild ends of the spectrum that have only been identified through NBS MCAD mutation c.199 C>T Never seen in patients picked up clinically For MCAD, biochemical parameters overlap between symptomatic and ‘probable mild’ cases, making decision-making about management challenging. Most clinics implement some type of treatment protocol. MCAD treatment is relatively benign though. Possibly in the future, there will be enough evidence that patients with 199C>T mutation are at very low risk of hypoglycemia.

23
Whom do we see? Patients who may not need any management
Disorders considered extremely rare but seen in large numbers via NBS programs Reported cases have significant morbidity NBS pickups are mostly mild 3MCC, SCAD Biochemical phenotype

24
Proceeding with Caution (Reasons to be Thoughtful)
Proceeding with caution Not screening Core diseases vs secondary targets/unintended targets What is reported vs withheld? Will we p/u untreatable conditions? What is the impact of false positives on families? No long-term outcome data – consider research paradigm Consider infrastructure needed for f/u Should abnormalities of unclear significance be reported or withheld? Harm may come to kids who are treated to ultimately don’t need to be. What about conditions that are not treatable? Consider expanding newborn screening within a research paradigm to create outcome data Consider the manpower and expertise needed to manage these families

25
Other Benefits to Screening
For disorders in which proven, effective treatment is not available, or very new. Consider non-medical benefits: Avoid the diagnostic odyssey Allow for reproductive decision making before future children are born Allow for early access to clinical trials for new therapies Emotional preparation for disease

26
What Are We Screening For?
9 OA 5 FAO 6 AA 3 Hb Pathies 6 Others CORE PANEL IVA GA I HMG MCD MUT 3MCC Cbl A,B PROP BKT MCAD VLCAD LCHAD TFP CUD PKU MSUD HCY CIT ASA TYR I Hb SS Hb S/ßTh Hb S/C CH BIOT CAH GALT HEAR CF

27
Cit/arg, cit/phe, cit/tyr, asa/arg
What Are We Measuring? Disorder 1° Marker 2° Marker Ratios VLCADD C14:1 C14,C16,C18,C18:1 C14:1/C16 LCHADD C16OH C14,C14:1,C16:C18,C18:1,C18:1OH, C18OH C16OH:C16 TFP CUD C0 All carnitines (low) PA C3 C2 C3/C0, C3/C2, C3/C16 MMAs C4DC IVA C5 C5/C2, C5/C0, C5/C3 GA-1 C5DC C5DC/C8, C5DC/C16, C5DC/C5OH BKT C5:1 C5OH C5OH/C8 HMG C6DC HCSD CIT Cit Cit/arg ASA Asa Cit/arg, cit/phe, cit/tyr, asa/arg TYR Suac Tyr
 PA. C3. C2. C3/C0, C3/C2, C3/C16. MMAs. C4DC. IVA. C5. C5/C2, C5/C0, C5/C3. GA-1. C5DC. C5DC/C8, C5DC/C16, C5DC/C5OH. BKT. C5:1. C5OH. C5OH/C8. HMG. C6DC. HCSD. CIT. Cit. Cit/arg. ASA. Asa. Cit/arg, cit/phe, cit/tyr, asa/arg. TYR. Suac. Tyr.")
28
Emma 13 months old Normal pregnancy and delivery Healthy
Normal eating pattern, no allergies or intolerances Feb 2008: Vomited Saturday and 4-5 times throughout the weekend No fever Sleeping for extended periods – parents concerned but previous fever had same pattern. Parents gave Pedialyte

29
Emma 4 ½ y brother, parents sick on Sunday/Monday. Same symptoms
Monday night 9:30 checked on E Raspy breathing – thought respiratory problem but not worried Tuesday morning 11am she was found motionless in her crib and pronounced dead at the scene

30
Emma Autopsy revealed fatty changes to liver
Coroner requested newborn screening blood spot be sent for acylcarnitine profile Diagnostic for Very Long Chain Acyl-Co A Dehydrogenase Deficiency (VLCAD)
")
31
VLCAD Disorder of long chain fatty acid breakdown C14, C14:1 C16, C18
Normal beta oxidation occurs in mitochondria

32
Fatty Acid Oxidation During times of fasting, fatty acids are primary substrate for energy production in liver, cardiac muscle and skeletal muscle Brain uses ketones (produced by normal b-oxidation)
")
33
Fatty Acid Oxidation With each pass through the cycle, the fatty acid is shortened by 2 carbon atoms as Acetyl-CoA, which is send to the TCA (tricarboxylic acid) cycle to be converted to energy . A fatty acid will cycle through as many times as necessary to generate these 2-carbon fragments. The enzymes along the path are somewhat specific for each chain length.
 cycle to be converted to energy . A fatty acid will cycle through as many times as necessary to generate these 2-carbon fragments. The enzymes along the path are somewhat specific for each chain length.")
34
VLCAD Enzyme VLCAD enzyme sits on inner mitochondrial membrane
Catalyzes first step of b-oxidation for C14-C20 Defect leads to impaired energy production during times of fasting stress Accumulation of toxic long-chain acyl-CoA intermediates within mitochondria Steatosis (fatty accumulation/degeneration) seen in hepatic, cardiac and skeletal muscle
 seen in hepatic, cardiac and skeletal muscle.")
35
VLCAD Presentations Hypertrophic cardiomyopathy, with hypoglycemia and skeletal myopathy, lethargy, failure to thrive Usually present birth-5 months Hypoglycemia, hepatomegaly, muscle weakness without cardiac manifestations Late infancy – older childhood Muscle weakness/pain, rhabdomyolysis with exercise or illness. No hypoglycemia or cardiac Teens to adulthood

36
VLCAD Treatment Diet low in long-chain fats (Portagen, Monogen = 87%, 90% of fats as MCT) Additional medium chain fats (MCT oil, walnut oil) Carnitine 100 mg/kg/day Avoidance of fasting Treating illness with IV glucose support

37
VLCAD Diagnosis Newborn screening Plasma acylcarnitine profile
Urine organic acids (should be normal) DNA sequencing VLCAD is included in the newborn screening program in all but two states now.
 DNA sequencing. VLCAD is included in the newborn screening program in all but two states now.")
38
Zach Testing Family referred to genetics by coroner
Parents requested testing for older brother Acylcarnitine ordered DNA sequencing of ACADVL gene ordered Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.

39
Normal acylcarnitine profile

40
Acylcarnitine – Zach 5 y.o.
C16:1- nl C14 C16 - nl Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.

41
Zach Testing Reported: mild elevation of C14 and C14:1 with low free carnitine. VLCAD cannot be ruled out Recommend supplementing with carnitine and retest in 1 week Family left for Disneyland DNA testing results back before AC repeat Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.

42
Zach Testing Zach’s DNA testing reveal he is affected.
Family seen in BCG clinic, started on treatment. Consent to obtain NBS blood spot obtained Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.

43
Acylcarnitine – Zach 5 y.o.
Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out. C14:1 C14 C16 - nl C16:1- nl C18 - nl

44
Acylcarnitine – Zach newborn
Cutoff is 0.65 C18 C16:1

45
Zach Clinical picture 5 y.o Healthy No symptoms of muscle weakness
CPK = 315U/L (35-230) No hepatomegaly AST= 49 (5-41) ALT= 23 Bilirubin conj, unconj = normal (0.0, 0.4) No evidence of cardiac involvement Has had several viral illnesses in his lifetime without difficulty Once on carnitine, AC profile was classic for VLCAD Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.
 No hepatomegaly. AST= 49 (5-41) ALT= 23. Bilirubin conj, unconj = normal (0.0, 0.4) No evidence of cardiac involvement. Has had several viral illnesses in his lifetime without difficulty. Once on carnitine, AC profile was classic for VLCAD. Reported: free carnitine low, mild elevation of C14 and C14:1, VLCAD can’t be ruled out.")
46
Newborn Screening – A Team Effort
The Presumptive Positive Phase DOH NBS Laboratory Personnel Mike Glass, Sheila Weiss, John Thompson, Carol-Nucup-Villaruz, Charlene Adams, Jessica Dolle, many laboratory technologists The Diagnostic Confirmation Phase CHRMC Diagnostic Laboratory Personnel Sihoun Hahn, Rhona Jack, Lisa Sniderman King, Cindy Gordon, Nancy McDowell Laura Mitchell, Diane Rebholz, Malcolm Reider Monica Jensen, Ngoc-Diep Pham, Min Zhang

47
Newborn Screening – A Team Effort
The Clinical Follow-up Phase Clinic Personnel All previously screened disorders: (PKU, MCAD, gal, btd, msud, hcys): UW: Ron Scott, Cris Trahms, Beth Ogata, Janie Heffernan, Jan Garretson, Stefanie Uhrich, Angie Fox All expanded screening disorders (FAOs, OAs…etc): CHRMC: Lawrence Merritt, Michael Raff, Sihoun Hahn, Sue Hale, Kelly McKean, Melissa Edwards, Lisa Sniderman King, Penny Schubert
: UW: Ron Scott, Cris Trahms, Beth Ogata, Janie Heffernan, Jan Garretson, Stefanie Uhrich, Angie Fox. All expanded screening disorders (FAOs, OAs…etc): CHRMC: Lawrence Merritt, Michael Raff, Sihoun Hahn, Sue Hale, Kelly McKean, Melissa Edwards, Lisa Sniderman King, Penny Schubert.")
48
What Happens After a Positive
NBS Lab notifies all clinical f/u and key laboratory personnel of referral. Laboratory technologists prioritize samples and collate results Multiple tests (AA, OA, AC) on each kiddo Interpreted together once all are completed Uniform, concise reporting
 on each kiddo. Interpreted together once all are completed. Uniform, concise reporting.")
49
2008 Cases 58 total cases since Jan
51 since expanded NBS started (July 21) 8 true positives for targeted disorders (MCAD and PKU) 1 true positive for secondary disorder (CblC) Elev C3 1° targets MMA/PA/CblA,B 38 FP/FPA – targeted disorder ruled out False Positive Active Persistent elevations in ‘normal’ baby Carriers (ie. further testing needed) Benign forms (D/G galactosemia) These are active because they require genetic counseling or lab repeats They are reclassified as FP when case is closed False Positive 1 mom with low free carnitine Several D/G galactosemia A few VLCAD carriers 10 pending (waiting for samples) 2 Unknown
 8 true positives for targeted disorders (MCAD and PKU) 1 true positive for secondary disorder (CblC) Elev C3 1° targets MMA/PA/CblA,B. 38 FP/FPA – targeted disorder ruled out. False Positive Active. Persistent elevations in ‘normal’ baby. Carriers (ie. further testing needed) Benign forms (D/G galactosemia) These are active because they require genetic counseling or lab repeats. They are reclassified as FP when case is closed. False Positive. 1 mom with low free carnitine. Several D/G galactosemia. A few VLCAD carriers. 10 pending (waiting for samples) 2 Unknown.")
50
2008 Cases True Positive False Positive Pending Unknown

Similar presentations

















>")

