Download presentation
Presentation is loading. Please wait.

1
Teresa K.Attwood School of Biological Sciences University of Manchester, Oxford Road Manchester M13 9PT, UK http://www.bioinf.man.ac.uk/dbbrowser/ Bioinformatics: gene-protein-structure-function

2
Foreword Predicting genes in uncharacterised genomic DNA is one of the main problems facing sequence annotators. De novo prediction methods (searching for splice-site consensus motifs, biased codon usage, etc.) have been only partially successful, & investigators have found that the surest way of predicting a gene is by alignment with a homologous protein sequence.
 have been only partially successful, & investigators have found that the surest way of predicting a gene is by alignment with a homologous protein sequence..")
3
Overview In silico structure &function prediction –the Holy Grail –a reality check What methods are available –PROSITE, PRINTS, Pfam, etc. Why not just use PSI-BLAST? Expert systems & other integrated approaches Conclusions

4
The Holy Grail of bioinformatics...to be able to understand the words in a sequence sentence that form a particular protein structure

5
The reality of sequence analysis...isn't so glamorous....but means we can recognise words that form characteristic patterns, even if we don't know the precise syntax to build complete protein sentences

6
Science fact & fiction The state of the art is pattern recognition Sequence pattern recognition is easier to achieve & more reliable than fold recognition –which is ~50% reliable even in expert hands Prediction is still not possible –& is unlikely to be so for decades to come (if ever) Structural genomics will yield representative structures for many (not all) proteins in future –structures of new sequences will be determined by modelling –prediction will become an academic exercise But, to debunk a popular myth, knowing structure alone does not inherently tell us function
 Structural genomics will yield representative structures for many (not all) proteins in future –structures of new sequences will be determined by modelling –prediction will become an academic exercise But, to debunk a popular myth, knowing structure alone does not inherently tell us function")
7
In silico function prediction …a reality check What is the function of this structure? What is the function of this sequence? What is the function of this motif? –the fold provides a scaffold, which can be decorated in different ways by different sequences to confer different functions - knowing the fold & function allows us to rationalise how the structure effects its function at the molecular level

8
What's in a sequence?

9
Full domain alignment methods Single motif methods Multiple motif methods Fuzzy regex (eMOTIF) Exact regex (PROSITE) Profiles (PROFILE LIBRARY) HMMs (Pfam) Identity matrices (PRINTS) Weight matrices (BLOCKS) Methods for family analysis
 Exact regex (PROSITE) Profiles (PROFILE LIBRARY) HMMs (Pfam) Identity matrices (PRINTS) Weight matrices (BLOCKS) Methods for family analysis")
10
The challenge of family analysis highly divergent family with single function? superfamily with many diverse functional families? –must distinguish if function analysis done in silico –a tough challenge!

11
In the beginning was PROSITE [GSTALIVMYWC]-[GSTANCPDE]-{EDPKRH}-X(2)-[LIVMNQGA]-X(2)-[LIVMFT]-[GSTANC]-LIVMFYWSTAC]-[DENH]-R TM domain
![In the beginning was PROSITE [GSTALIVMYWC]-[GSTANCPDE]-{EDPKRH}-X(2)-[LIVMNQGA]-X(2)-[LIVMFT]-[GSTANC]-LIVMFYWSTAC]-[DENH]-R TM domain](http://images.slideplayer.com/26/8872913/slides/slide_11.jpg "In the beginning was PROSITE [GSTALIVMYWC]-[GSTANCPDE]-{EDPKRH}-X(2)-[LIVMNQGA]-X(2)-[LIVMFT]-[GSTANC]-LIVMFYWSTAC]-[DENH]-R TM domain")
12
Diagnostic limitations of PROSITE G_PROTEIN_RECEPTOR; PATTERN PS00237; G-protein coupled receptor signature [GSTALIVMYWC]-[GSTANCPDE]-{EDPKRH}-X(2)-[LIVMNQGA]- X(2)-[LIVMFT]-[GSTANC]-[LIVMFYWSTAC]-[DENH]-R /TOTAL=1121(1121); /POS=1057(1057); /FALSE_POS=64(64); /FALSE_NEG=112; /PARTIAL=48; UNKNOWN=0(0) This represents an apparent 20% error rate –the actual rate is probably higher Thus, a match to a pattern is not necessarily true –& a mis-match is not necessarily false! False-negatives are a fundamental limitation to this type of pattern matching –if you don't know what you're looking for, you'll never know you missed it!
![Diagnostic limitations of PROSITE G_PROTEIN_RECEPTOR; PATTERN PS00237; G-protein coupled receptor signature [GSTALIVMYWC]-[GSTANCPDE]-{EDPKRH}-X(2)-[LIVMNQGA]- X(2)-[LIVMFT]-[GSTANC]-[LIVMFYWSTAC]-[DENH]-R /TOTAL=1121(1121); /POS=1057(1057); /FALSE_POS=64(64); /FALSE_NEG=112; /PARTIAL=48; UNKNOWN=0(0) This represents an apparent 20% error rate –the actual rate is probably higher Thus, a match to a pattern is not necessarily true –& a mis-match is not necessarily false.](http://images.slideplayer.com/26/8872913/slides/slide_12.jpg "False-negatives are a fundamental limitation to this type of pattern matching –if you don t know what you re looking for, you ll never know you missed it!.")
13
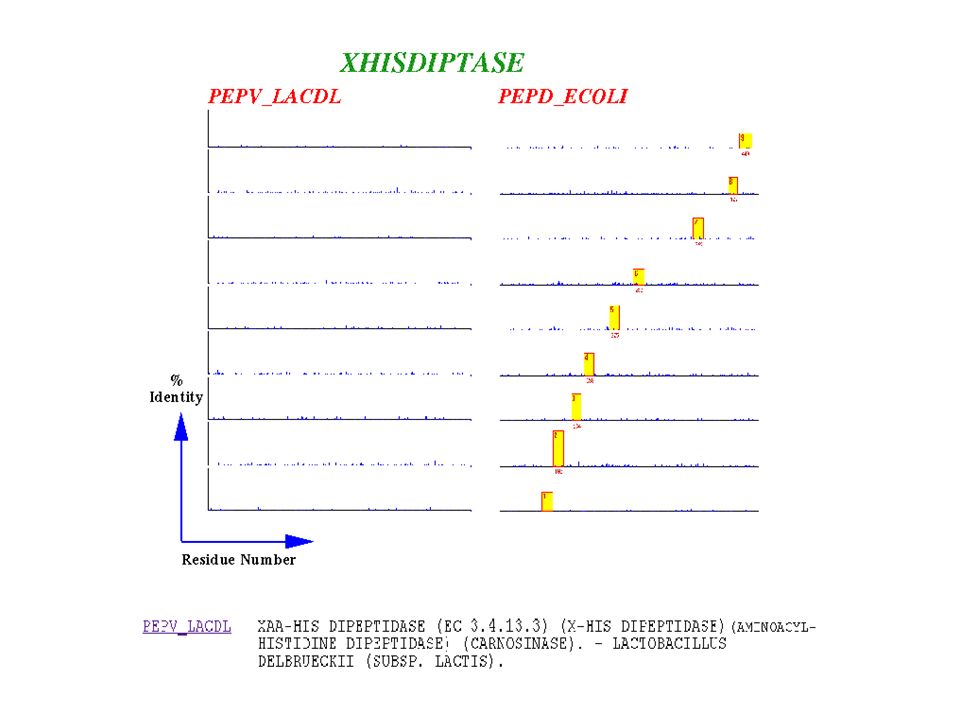
TM domain loop region Then came PRINTS

14
loop region TM domain Hierarchical family analysis

15
What is PRINTS? (not the best thing since sliced bread, but....) A db of diagnostic fingerprints that characterise proteins –family analysis is hierarchical, allowing fine-grained diagnoses Fingerprints are groups of conserved motifs, used for iterative db searching –iteration refines the fingerprint –potency is gained from the mutual context of motif neighbours –results are biologically more meaningful than from single motifs –results are manually annotated prior to inclusion in the db PRINTS has many applications, e.g.: –basis of BLOCKS & eMOTIF –EditToTrEMBL - to annotate TrEMBL –provide annotation & hierarchical protein classification in InterPro
 A db of diagnostic fingerprints that characterise proteins –family analysis is hierarchical, allowing fine-grained diagnoses Fingerprints are groups of conserved motifs, used for iterative db searching –iteration refines the fingerprint –potency is gained from the mutual context of motif neighbours –results are biologically more meaningful than from single motifs –results are manually annotated prior to inclusion in the db PRINTS has many applications, e.g.: –basis of BLOCKS & eMOTIF –EditToTrEMBL - to annotate TrEMBL –provide annotation & hierarchical protein classification in InterPro.")
19
N C Visualising fingerprints

20
Diagnosing partial matches

25
-opioid receptor -opioid receptor -opioid receptor true

26
Why bother with family dbs? Seq searches won't always allow outright diagnosis –BLAST & FASTA are not infallible & often can't assign significant scores –outputs may be complicated by the multi-domain or modular nature of the protein, compositionally biased regions, repeats & so on –annotations of retrieved hits may be incorrect Pattern dbs contain potent descriptors –so, distant relationships missed by pairwise search tools may be captured by one or more of the family or functional site distillations

27
Overview of resources PROSITE (SIB) - 1144 entries –single motifs (regexs) - best with small highly conserved sites Profile library (ISREC) - ~300 entries –weight matrices - good with divergent domains & superfamilies PRINTS (Manchester) - 1750 entries –multiple motifs (fingerprints) - best for families and sub-families Pfam (Sanger Centre) - 3849 entries –HMMs - good with divergent domains & superfamilies Blocks (FHCRC) - ~2608 entries –multiple motifs (derived from InterPro & PRINTS) eMOTIF (Stanford) –permissive regexs (derived from PRINTS & BLOCKS)
 entries –single motifs (regexs) - best with small highly conserved sites Profile library (ISREC) - ~300 entries –weight matrices - good with divergent domains & superfamilies PRINTS (Manchester) entries –multiple motifs (fingerprints) - best for families and sub-families Pfam (Sanger Centre) entries –HMMs - good with divergent domains & superfamilies Blocks (FHCRC) - ~2608 entries –multiple motifs (derived from InterPro & PRINTS) eMOTIF (Stanford) –permissive regexs (derived from PRINTS & BLOCKS)")
28
Designing a search protocol Given a newly-determined sequence, want to know –what is my protein? –to what family does it belong? –what is its function? –how can we explain this in structural terms? Given the variety of dbs available, rather than rely on just one, it is important to devise a search protocol –search the sequence & family dbs –estimate significance - compare results & find a consensus

29
This does not simply mean.... BLAST + PROSITE (e.g., on the Web) –or FASTA + motifs/profiles (e.g., using GCG) But this is still what most people do –including so-called expert systems for genome analysis
 –or FASTA + motifs/profiles (e.g., using GCG) But this is still what most people do –including so-called expert systems for genome analysis.")
30
Expert systems for functional analysis...from genome data to biological knowledge GeneQuiz- Automatic protein function annotation MAGPIE- Automatic genome analysis PEDANT- Automatic analysis of proteins How they describe themselves: GeneQuiz- Expert system for derivation of functional information MAGPIE- Automated Genome Project Investigation Environment PEDANT- Complete functional & structural characterisation of protein sequences What they do: GeneQuiz- BLAST/FASTA, PROSITE, BLOCKS MAGPIE- BLAST/FASTA, PROSITE, BLOCKS PEDANT- BLAST/FASTA, PROSITE, BLOCKS

31
D10226R13F63 UL78_HCMVA R05H51 450320 Challenges for expert systems

32
* * *

33
What GeneQuiz said… a thrombin receptor?

34
What GeneQuiz said later… *

35
Other integrated approaches The European InterPro project To simplify sequence analysis, the family databases are being integrated to create a unified annotation resource – InterPro –current release has 5312 entries –a central annotation resource, with pointers to its satellite dbs –initial partners were PRINTS, PROSITE, profiles & Pfam –new partners include ProDom, TIGRfam, SMART & hopefully others (e.g., BLOCKS, MetaFam) –lags behind its sources –major role in fly & human genome annotation
 –lags behind its sources –major role in fly & human genome annotation")
36
InterPro – method comparison

37
Ground rules for bioinformatics Don't always believe what programs tell you –they're often misleading & sometimes wrong! Don't always believe what databases tell you –they're often misleading & sometimes wrong! Don't always believe what lecturers tell you –they're sometimes misleading & often wrong! In short, don't be a naive user –when computers are applied to biology, it is vital to understand the difference between mathematical & biological significance –computers don’t do biology –they do sums –quickly!

38
Conclusions Success of search protocols based only on BLAST & PROSITE is likely to be limited –beware ‘expert’ systems –understand the methods No db is best - use several –different methods provide different perspectives –dbs aren’t complete & their contents don’t fully overlap The more dbs searched, harder to interpret results –hence s/w being designed to give "intelligent" consensus outputs The more computers are used to automate analysis, the greater the need for collaboration –between s/w developers, annotators & ‘wet’ experimentalists Long way from having reliable analytical tools –but with the right approach…

Similar presentations







>")









: Look at the Swiss-Prot annotation (in a random ‘glycosylated’ entry)>")
>")



 –Fold recognition (threading) Outstanding difficult.>")
 a text editor (Notepad.>")

