Download presentation
Presentation is loading. Please wait.

1
Applied Bioinformatics Week 3

2
Theory I Similarity Dot plot

3
3 Introduction to Bioinformatics http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: SEQUENCE ALIGNMENT http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm 3.2 On sequence alignment Sequence alignment is the most important task in bioinformatics!

4
4 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: SEQUENCE ALIGNMENT 3.2 On sequence alignment Sequence alignment is important for: * prediction of function * database searching * gene finding * sequence divergence * sequence assembly

5
5 http:// www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http:// www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: SEQUENCE ALIGNMENT 3.3 On sequence similarity Homology: genes that derive from a common ancestor-gene are called homologs Orthologous genes are homologous genes in different organisms Paralogous genes are homologous genes in one organism that derive from gene duplication Gene duplication: one gene is duplicated in multiple copies that therefore free to evolve and assume new functions

6
6 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm HOMOLOGOUS and PARALOGOUS

7
7 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm HOMOLOGOUS and PARALOGOUS

8
8 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm HOMOLOGOUS and PARALOGOUS versus ANALOGOUS

9
? globin plants Ath-g analogs

10
10 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: SEQUENCE ALIGNMENT: sequence similarity Causes for sequence (dis)similarity mutation: a nucleotide at a certain location is replaced by another nucleotide (e.g.: A T A → A G A) insertion: at a certain location one new nucleotide is inserted inbetween two existing nucleotides (e.g.: AA → A G A) deletion: at a certain location one existing nucleotide is deleted (e.g.: AC T G → AC - G) indel: an in sertion or a del etion
similarity mutation: a nucleotide at a certain location is replaced by another nucleotide (e.g.: A T A → A G A) insertion: at a certain location one new nucleotide is inserted inbetween two existing nucleotides (e.g.: AA → A G A) deletion: at a certain location one existing nucleotide is deleted (e.g.: AC T G → AC - G) indel: an in sertion or a del etion")
11
Similarity We can only measure current similarity We can form hypothesi

12
Similarity Searching DotPlot Needleman-Wunsch Smith-Waterman FASTA BLAST

13
Dot Plot Writing one sequence horizontally Writing the other vertically At each intersection with equal nucleotides make a dot in the matrix

14
Dot Plot

15
Messy? Strong similarities can be visually enhanced Select a window size and a similarity score for that window (e.g. 10 and 8) Create a new matrix with dots where the window score >= 8
 Create a new matrix with dots where the window score >= 8.")
16
Dot Plot

17
Dot Plot Interpretation

18
Creating a Dot Plot

19
End Theory I Mindmapping 10 min break

20
Practice I Dot plot

21
Dot Plot ACGTGTGCGTTTGAAC GGGTGTTCGTTTAAAC Make a Dot plot for the two sequences above Use a window of 3 to refine the view Can you use Excel? Get any two DNA sequences and try the tool below –http://www.vivo.colostate.edu/molkit/dnadot/http://www.vivo.colostate.edu/molkit/dnadot/

22
Similarity Searching DotPlot Needleman-Wunsch Smith-Waterman FASTA BLAST

23
Definitions Optimal alignment - one that exhibits the most correspondences. It is the alignment with the highest score. May or may not be biologically meaningful. Global alignment - Needleman-Wunsch (1970) maximizes the number of matches between the sequences along the entire length of the sequences. Local alignment - Smith-Waterman (1981) gives the highest scoring local match between two sequences.
 maximizes the number of matches between the sequences along the entire length of the sequences. Local alignment - Smith-Waterman (1981) gives the highest scoring local match between two sequences..")
24
How can we find an optimal alignment? ACGTCTGATACGCCGTATAGTCTATCT CTGAT---TCG-CATCGTC--T-ATCT How many possible alignments? C(27,7) gap positions = ~888,000 possibilities Dynamic programming: The Needleman & Wunsch algorithm 1 27
 gap positions = ~888,000 possibilities Dynamic programming: The Needleman & Wunsch algorithm")
25
Time Complexity Consider two sequences: AAGT AGTC How many possible alignments the 2 sequences have? 2nn = (2n)!/(n!) 2 = (2 2n / n ) = (2 n ) = 70
!/(n!) 2 = (2 2n / n ) = (2 n ) = 70.")
26
Scoring a sequence alignment Match/mismatch score:+1/+0 Open/extension penalty:–2/–1 ACGTCTGATACGCCGTATAGTCTATCT ||||| ||| || |||||||| ----CTGATTCGC---ATCGTCTATCT Matches: 18 × (+1) Mismatches: 2 × 0 Open: 2 × (–2) Extension: 5 × (–1) Score = +9
 Mismatches: 2 × 0 Open: 2 × (–2) Extension: 5 × (–1) Score = +9")
27
Pairwise Global Alignment Computationally: –Given: a pair of sequences (strings of characters) –Output: an alignment that maximizes the similarity
 –Output: an alignment that maximizes the similarity")
28
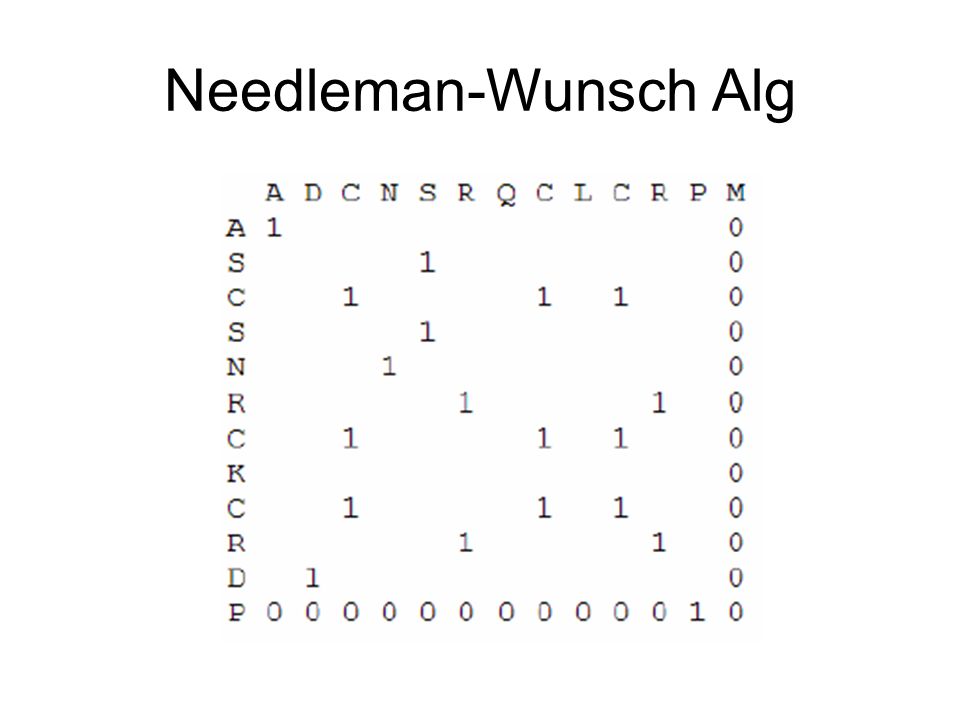
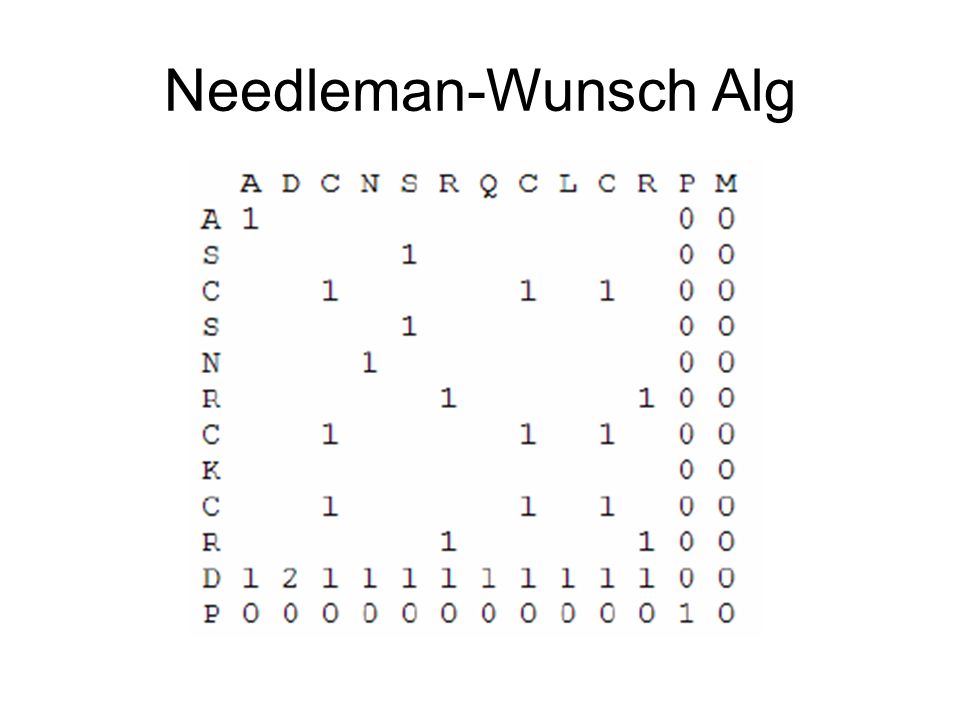
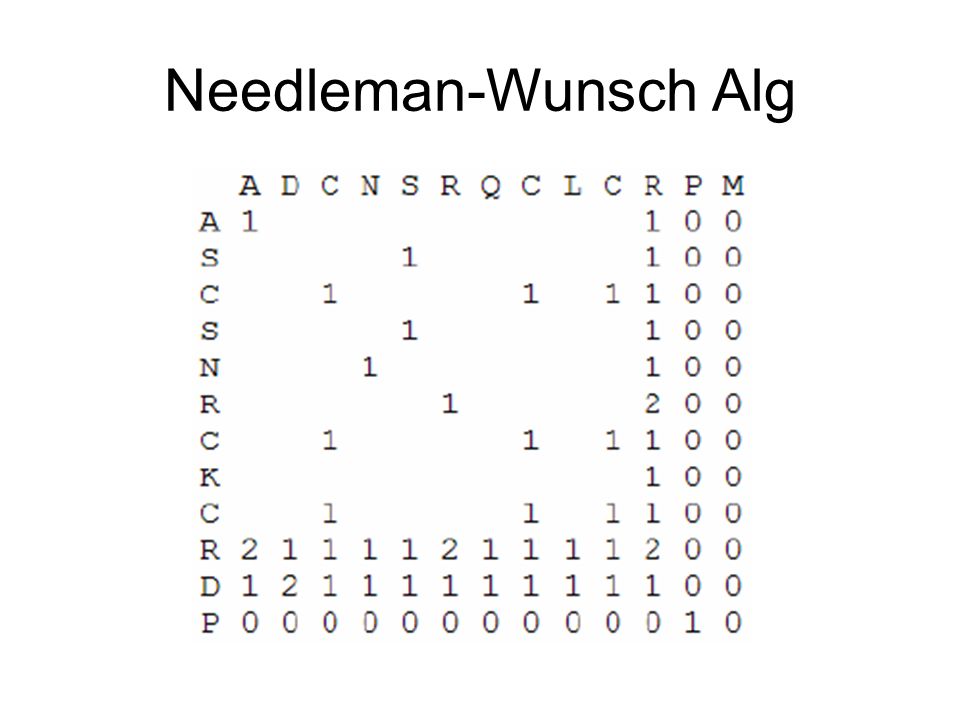
Needleman-Wunsch Alg

36
Which Alignment is better? For scoring use: –Match 1 –Mismatch 0 –Gap open -2 –Gap extension -1 How can substitution matrices be integrated?

37
Needleman & Wunsch Place each sequence along one axis Place score 0 at the up-left corner Fill in 1 st row & column with gap penalty multiples Fill in the matrix with max value of 3 possible moves: –Vertical move: Score + gap penalty –Horizontal move: Score + gap penalty –Diagonal move: Score + match/mismatch score The optimal alignment score is in the lower-right corner To reconstruct the optimal alignment, trace back where the max at each step came from, stop when hit the origin.

38
Three steps in Needleman-Wunsch Algorithm Initialization Scoring Trace back (Alignment) Consider the two DNA sequences to be globally aligned are: ATCG (x=4, length of sequence 1) TCG (y=3, length of sequence 2) Pooja Anshul Saxena, University of Mississippi
 Consider the two DNA sequences to be globally aligned are: ATCG (x=4, length of sequence 1) TCG (y=3, length of sequence 2) Pooja Anshul Saxena, University of Mississippi")
39
Scoring Scheme Match Score = +1 Mismatch Score = -1 Gap penalty = -1 Substitution Matrix ACGT A1 C 1 G 1 T 1 Pooja Anshul Saxena, University of Mississippi

40
Initialization Step Create a matrix with X +1 Rows and Y +1 Columns The 1st row and the 1st column of the score matrix are filled as multiple of gap penalty TCG 0-2-3 A T-2 C-3 G-4 Pooja Anshul Saxena, University of Mississippi

41
Scoring The score of any cell C(i, j) is the maximum of: scorediag = C(i-1, j-1) + S(i, j) = -1 scoreup = C(i-1, j) + g = -2 scoreleft = C(i, j-1) + g = -2 where S(i, j) is the substitution score for letters i and j, and g is the gap penalty TCG 0 -2-3 A T-2 C-3 G-4 Max -> C(i,j) S(T,A)
 is the maximum of: scorediag = C(i-1, j-1) + S(i, j) = -1 scoreup = C(i-1, j) + g = -2 scoreleft = C(i, j-1) + g = -2 where S(i, j) is the substitution score for letters i and j, and g is the gap penalty TCG A T-2 C-3 G-4 Max -> C(i,j) S(T,A)")
42
Scoring …. Example: The calculation for the cell C(2, 2): scorediag = C(i-1, j-1) + S(I, j) = 0 + -1 = -1 scoreup = C(i-1, j) + g = -1 + -1 = -2 scoreleft = C(i, j-1) + g = -1 + -1 = -2 TCG 0-2-3 A T-2 C-3 G-4 Pooja Anshul Saxena, University of Mississippi
: scorediag = C(i-1, j-1) + S(I, j) = = -1 scoreup = C(i-1, j) + g = = -2 scoreleft = C(i, j-1) + g = = -2 TCG A T-2 C-3 G-4 Pooja Anshul Saxena, University of Mississippi.")
43
Scoring …. Final Scoring Matrix TCG 0-2-3 A -2-3 T-20-2 C-310 G-4-202 Pooja Anshul Saxena, University of Mississippi

44
Trace back The trace back step determines the actual alignment(s) that result in the maximum score There are likely to be multiple maximal alignments Trace back starts from the last cell, i.e. position X, Y in the matrix Gives alignment in reverse order Pooja Anshul Saxena, University of Mississippi

45
Trace back …. There are three possible moves: diagonally (toward the top-left corner of the matrix), up, or left Trace back takes the current cell and looks to the neighbor cells that could be direct predecessors. This means it looks to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). The algorithm for trace back chooses as the next cell in the sequence one of the possible predecessors Pooja Anshul Saxena, University of Mississippi
, up, or left Trace back takes the current cell and looks to the neighbor cells that could be direct predecessors. This means it looks to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). The algorithm for trace back chooses as the next cell in the sequence one of the possible predecessors Pooja Anshul Saxena, University of Mississippi.")
46
Trace back …. The only possible predecessor is the diagonal match/mismatch neighbor. If more than one possible predecessor exists, any can be chosen. This gives us a current alignment of Seq 1: G | Seq 2: G TCG 0-2-3 A -2-3 T-20-2 C-310 G-4-202 Pooja Anshul Saxena, University of Mississippi

47
Trace back …. Final Trace back Best Alignment: A T C G | | | | _ T C G TCG 0-2-3 A -2-3 T-20-2 C-310 G-4-202 Pooja Anshul Saxena, University of Mississippi

48
Similarity Searching DotPlot Needleman-Wunsch Smith-Waterman FASTA BLAST

49
Local Alignment Problem first formulated: –Smith and Waterman (1981) Problem: –Find an optimal alignment between a substring of s and a substring of t Algorithm: – is a variant of the basic algorithm for global alignment
 Problem: –Find an optimal alignment between a substring of s and a substring of t Algorithm: – is a variant of the basic algorithm for global alignment")
50
Motivation Searching for unknown domains or motifs within proteins from different families –Proteins encoded from Homeobox genes (only conserved in 1 region called Homeo domain – 60 amino acids long) –Identifying active sites of enzymes Comparing long stretches of anonymous DNA Querying databases where query word much smaller than sequences in database Analyzing repeated elements within a single sequence
 –Identifying active sites of enzymes Comparing long stretches of anonymous DNA Querying databases where query word much smaller than sequences in database Analyzing repeated elements within a single sequence")
51
Smith-Waterman Alg Very similar to Needleman-Wunsch Determines local instead of global alignment Scores can drop and increase Alignments are calculated between 0 and 0 scores

52
Three steps in Smith-Waterman Algorithm Initialization Scoring Trace back (Alignment) Consider the two DNA sequences to be globally aligned are: ATCG (x=4, length of sequence 1) TCG (y=3, length of sequence 2) Pooja Anshul Saxena, University of Mississippi
 Consider the two DNA sequences to be globally aligned are: ATCG (x=4, length of sequence 1) TCG (y=3, length of sequence 2) Pooja Anshul Saxena, University of Mississippi")
53
Scoring Scheme Match Score = +1 Mismatch Score = -1 Gap penalty = -1 Substitution Matrix ACGT A1 C 1 G 1 T 1 Pooja Anshul Saxena, University of Mississippi

54
Initialization Step Create a matrix with X +1 Rows and Y +1 Columns The 1st row and the 1st column of the score matrix are filled with 0s TCG 0000 A0 T0 C0 G0 Pooja Anshul Saxena, University of Mississippi

55
Scoring The score of any cell C(i, j) is the maximum of: scorediag = C(i-1, j-1) + S(I, j) scoreup = C(i-1, j) + g scoreleft = C(i, j-1) + g And 0 (here S(I, j) is the substitution score for letters i and j, and g is the gap penalty) Pooja Anshul Saxena, University of Mississippi
 is the maximum of: scorediag = C(i-1, j-1) + S(I, j) scoreup = C(i-1, j) + g scoreleft = C(i, j-1) + g And 0 (here S(I, j) is the substitution score for letters i and j, and g is the gap penalty) Pooja Anshul Saxena, University of Mississippi")
56
Scoring …. Example: The calculation for the cell C(2, 2): scorediag = C(i-1, j-1) + S(I, j) = 0 + -1 = -1 scoreup = C(i-1, j) + g = 0 + -1 = -1 scoreleft = C(i, j-1) + g = 0 + -1 = -1 TCG 0000 A00 T0 C0 G0 Pooja Anshul Saxena, University of Mississippi
: scorediag = C(i-1, j-1) + S(I, j) = = -1 scoreup = C(i-1, j) + g = = -1 scoreleft = C(i, j-1) + g = = -1 TCG 0000 A00 T0 C0 G0 Pooja Anshul Saxena, University of Mississippi.")
57
Scoring …. Final Scoring Matrix Note: It is not mandatory that the last cell has the maximum alignment score! TCG 0000 A0000 T0100 C0021 G0013 Pooja Anshul Saxena, University of Mississippi

58
Trace back The trace back step determines the actual alignment(s) that result in the maximum score There are likely to be multiple maximal alignments Trace back starts from the cell with maximum value in the matrix Gives alignment in reverse order Pooja Anshul Saxena, University of Mississippi
 that result in the maximum score There are likely to be multiple maximal alignments Trace back starts from the cell with maximum value in the matrix Gives alignment in reverse order Pooja Anshul Saxena, University of Mississippi")
59
Trace back …. There are three possible moves: diagonally (toward the top-left corner of the matrix), up, or left Trace back takes the current cell and looks to the neighbor cells that could be direct predecessors. This means it looks to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). The algorithm for trace back chooses as the next cell in the sequence one of the possible predecessors. This continues till cell with value 0 is reached. Pooja Anshul Saxena, University of Mississippi
, up, or left Trace back takes the current cell and looks to the neighbor cells that could be direct predecessors. This means it looks to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). The algorithm for trace back chooses as the next cell in the sequence one of the possible predecessors. This continues till cell with value 0 is reached. Pooja Anshul Saxena, University of Mississippi.")
60
Trace back …. The only possible predecessor is the diagonal match/mismatch neighbor. If more than one possible predecessor exists, any can be chosen. This gives us a current alignment of Seq 1: G | Seq 2: G TCG 0000 A0000 T0100 C0021 G0013 Pooja Anshul Saxena, University of Mississippi

61
Trace back …. Final Trace back Best Alignment: T C G | | | T C G TCG 0000 A0000 T0100 C0021 G0013 Pooja Anshul Saxena, University of Mississippi

62
62 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: GLOBAL ALIGNMENT

63
63 http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm http://www.personeel.unimaas.nl/Westra/Education/BioInf/slides_of_bioinformatics.htm LECTURE 3: GLOBAL ALIGNMENT

64
Significance of Sequence Alignment Consider randomly generated sequences. What distribution do you think the best local alignment score of two sequences of sample length should follow? 1.Uniform distribution 2.Normal distribution 3.Binomial distribution (n Bernoulli trails) 4.Poisson distribution (n , np= ) 5.others
 4.Poisson distribution (n , np= ) 5.others.")
65
Extreme Value Distribution Y ev = exp(- x - e -x )
")
66
Extreme Value Distribution vs. Normal Distribution

67
“Twilight Zone” Some proteins with less than 15% similarity have exactly the same 3-D structure while some proteins with 20% similarity have different structures. Homology/non-homology is never granted in the twilight zone.

68
End of Theoretical Part 2 Mindmapping 10 min break

69
Needleman-Wunsch ACGTGTGCGTTTGAAC GGGTGTAGTCGTTTAAAC Apply the Needleman-Wunsch algorithm to these two sequences Score the alignments

70
Alignments Explanation for alignment algorithms –http://baba.sourceforge.net/http://baba.sourceforge.net/ Alignment of 2 sequences –http://www.expasy.org/tools/sim-prot.htmlhttp://www.expasy.org/tools/sim-prot.html Get any two amino acid sequences and try –http://bioinformatics.iyte.edu.tr/SmithWaterman/http://bioinformatics.iyte.edu.tr/SmithWaterman/

Similar presentations

















 Lecture 3: Pair-wise alignment Centre for Integrative Bioinformatics VU.>")





