Download presentation
Presentation is loading. Please wait.

1
Amyotrophic Lateral Sclerosis (ALS; Lou Gehrig’s disease)
")
2
Lou Gehrig

3
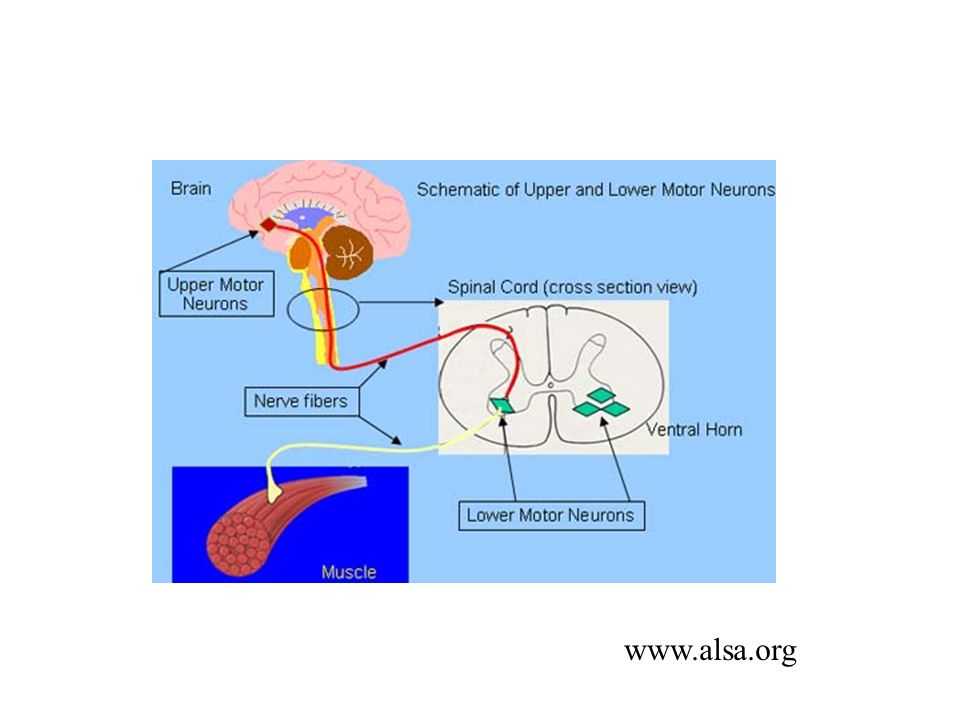
ALS is a “motor- neuron” disease, a neurodegenerative disease characterized by the selective death/degeneration of upper motor neurons (in the brain) and lower motor neurons (in the spinal cord). The upper motor neurons normally send the signals to the lower motor neurons, which send signals to muscles.

4
www.alsa.org
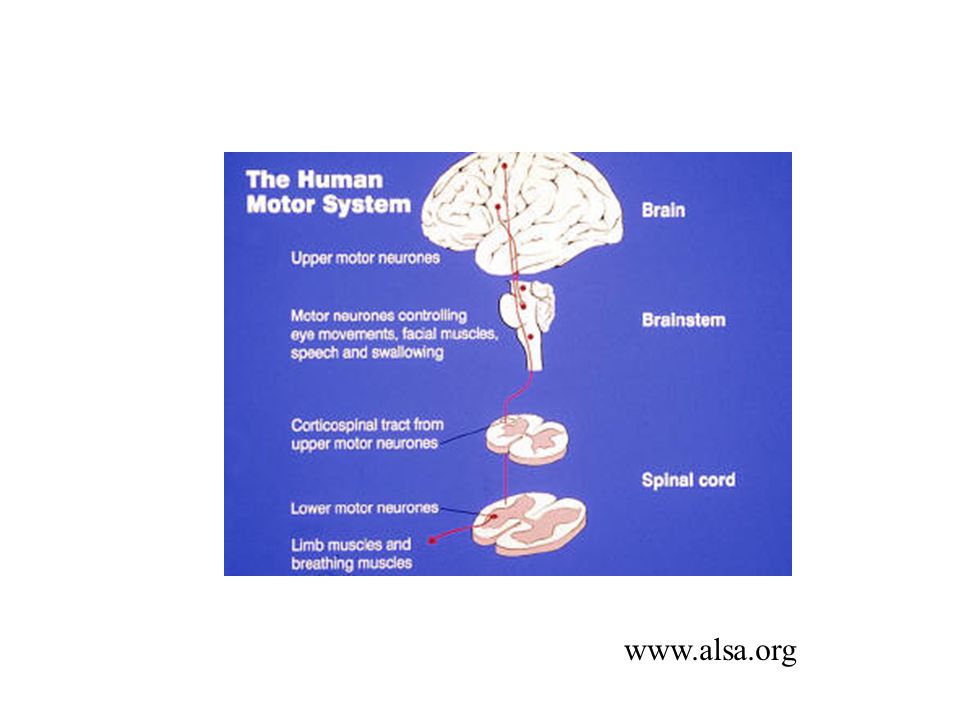
6
A-myo-trophic Lateral Sclerosis Amyotrophic: no muscle nourishment Lateral: refers to the the areas in a person's spinal cord where portions of the nerve cells that signal and control the muscles are located Sclerosis: scarring of the affected nerves

7
Degeneration of motor neurons in the spinal cord and brainstem results in degeneration of pyramidal tracts and severe atrophy of anterior spinal roots which is demonstrated here. Severe atrophy of anterior spinal roots in motorneuron disease

8
Atrophic muscle fibers in ALS patients

9
Muscular atrophy in ALS

10
Symptoms Earliest signs: 1-twitching (fasciculation), stiffness, cramping 2-weakness of the arms and of the legs. This results in an increased frequency of stumbling on uneven pavement or difficulty in climbing stairs. Arm weakness may lead to difficulty in grasping or holding a cup, for instance, or loss of coordination in fingers 3-more rarely, weakness of the muscles of the mouth. This results in difficulties for the patient to chew, swallow and speak. Later signs: The progression of the disease is accompanied by weight loss, fatigue, exaggerated reflexes, and decreased coordination. Ultimately, patients cannot walk, stand, eat, or breathe without assistance. Increased susceptibility to pneumonia and respiratory failure causes half to die within three years. However, muscles that controls eye movements and urinary sphincters are spared.

11
Diagnosis of ALS -No biological marker has been identified yet. -Series of clinical and neurological exams -MRI -myelogram of cervical spine (an x-ray analysis that allows the detection of lesions in selected area of the spinal cord) -muscle and/or nerve biopsy -electromyography (EMG) and nerve conduction velocity (NCV), to measure muscle response to nervous stimulation.
 -muscle and/or nerve biopsy -electromyography (EMG) and nerve conduction velocity (NCV), to measure muscle response to nervous stimulation..")
12
MRI in Amyotrophic Lateral Sclerosis (ALS)
")
13
Electromyography (EMG) A needle electrode is inserted through the skin into the muscle. Each muscle fiber that contracts will produce an action potential. The presence, size, and shape of the wave form of the action potential produced on the oscilloscope, provides information about the ability of the muscle to respond to nervous stimulation.

14
Epidemiology of ALS Incidence: 1-2 in 100,000 each year. At the moment in the U.S. there are 25,000 people affected by the disease. Median age of onset is 55 years old. Gender-related incidence: female:male ratio is 4:5. 10% of the cases are inherited, familial cases (FALS), whereas the 90% of the cases are sporadic (SALS) The life-span of a patient affected by ALS is 3 to 5 years, after the diagnosis.
, whereas the 90% of the cases are sporadic (SALS) The life-span of a patient affected by ALS is 3 to 5 years, after the diagnosis..")
15
Genetic of ALS

16
Toxic mechanisms in ALS

17
Is ALS a mitochondrial disease?

18
Evidences of mitochondrial dysfunction in ALS: -mitochondria aggregate in skeletal muscle and intramuscular nerve -mitochondria show abnormalities in proximal axons and nerves of the anterior horn of spinal cord -increased mitochondrial volume and calcium level -dysfunction of mitochondrial complex I and IV Decreased ATP production

19
Syklos et al., Calcium deposits in mitochondria of ALS patients CONTROL ALS

20
Superoxide Dismutase 1 SOD1 SOD1 is a ubiquitous mostly cytosolic protein SOD1 is comprised of 153 aa with an approximate molecular weight of 16kDa and is an active homodimer Each of the two dimers of SOD1 binds a Cu ++ and a Zn ++ ion. The reduction of Cu ++ to Cu + is behind the mechanism of SOD1 in regulating the dismutation of superoxide ion O 2 -. into hydrogen peroxide H 2 O 2 Cu 2+ + O 2-· Cu + + O 2 Cu + + O 2-· + 2H + Cu 2+ + H 2 O 2 2 O 2-· + 2H + H 2 O 2 + O 2 A catalase will subsequently reduce hydrogen peroxide to water.

21
SOD1 structure: the functional homodimer

22
Most common mutations in SOD1 related FALS

23
SOD1 and FALS -More than 125 mutations have been found on the SOD1 gene, 114 are related to ALS, most of them are missense mutations, only 12 are nonsense mutations or deletion mutants. -Most mutations reduce dismutation activity, however others retain full dismutase activity, still are related to the disease. Moreover, there is NO CLEAR CORRELATION between enzyme activity and progression of the disease. -In addition, in animal models, gene KO for SOD1 does not cause motorneuron disease, whereas overexpression of SOD1 does. In this respect, the simple manipulation of SOD1 dismutase activity IS NOT necessarily behind ALS/motorneuron disease.

24
SOD1 misfolds and aggregates in FALS SOD1A4V motorneurons SEDI Ab SEDI-reactive SOD1SOD1

25
Possible roles of mutated SOD1 in FALS Loss of physiological function: impaired dismutase activity Gain of toxic function: 1) Aberrant redox chemistry, probably due to changes in the conformation of SOD1, that leave the channel (the portion of the molecule accepting superoxide ion, i.e.) able to accept larger molecules. This can lead to peroxidation, tyrosine nitrosylation and reverse catalysis (due to improper binding of Zn ++ to the molecule that leads to formation of superoxide ion rather than dismutase activity). These activities are not a characteristic of ALL SOD1 mutations, thus remain partially controversial. 2) Protein instability and SOD1 aggregation. These activities are characteristic of all SOD1 mutants.
. These activities are not a characteristic of ALL SOD1 mutations, thus remain partially controversial. 2) Protein instability and SOD1 aggregation. These activities are characteristic of all SOD1 mutants..")
26
O 1 2 3 4 O-O…CuZn WT Peroxidation H H O Cu Zn.OH CuZn ONOO NO-Tyr Tyrosine Nitration Cu Zn toxic Cu, Zn Toxicity Aggregation Mutant SOD1 may mediate cytotoxic reactions involving: Mutant SOD1 may mediate cytotoxic reactions involving: 1) Copper catalysis/Zn-mediated toxicity 2) Protein aggregation O 1 2 3 4 O-O…CuZn WT Peroxidation H H O Cu Zn.OH CuZn ONOO NO-Tyr Tyrosine Nitration Cu Zn toxic Cu, Zn Toxicity Aggregation Mutant SOD1 may mediate cytotoxic reactions involving: Mutant SOD1 may mediate cytotoxic reactions involving: 1) Copper catalysis/Zn-mediated toxicity 2) Protein aggregation P.Pasinelli
 Copper catalysis/Zn-mediated toxicity 2) Protein aggregation O O-O…CuZn WT Peroxidation H H O Cu Zn.OH CuZn ONOO NO-Tyr Tyrosine Nitration Cu Zn toxic Cu, Zn Toxicity Aggregation Mutant SOD1 may mediate cytotoxic reactions involving: Mutant SOD1 may mediate cytotoxic reactions involving: 1) Copper catalysis/Zn-mediated toxicity 2) Protein aggregation P.Pasinelli")
27
Models of mutant SOD1-mediated toxicity

28
1- Formation of small and large aggregates that may impairs proteasomal activity. This would result in lack of proper degradation of different proteins including toxic mutant SOD1. 2- Sequestration within the aggregate of proteins that are important for the cell, like heat shock proteins (HSP70), thus impairing the physiological “protective” activity of these proteins. 3-SOD1 can sequester into aggregates the anti-apoptotic Bcl2. 4- Formation of SOD1 aggregates can be related to mitochondrial dysfunction and apoptosis. How SOD1 aggregates could be toxic?
, thus impairing the physiological protective activity of these proteins. 3-SOD1 can sequester into aggregates the anti-apoptotic Bcl2. 4- Formation of SOD1 aggregates can be related to mitochondrial dysfunction and apoptosis. How SOD1 aggregates could be toxic .")
29
SOD1 aggregates: mitochondrial dysfunction and apoptosis Studies in FALS

30
SOD1 associates with Bcl2, but not with Bax, in vitro… Exogenous SOD1Endogenous SOD1

31
…and in vivo miceHuman spinal cord

32
Bcl2 binds aggregated SOD1 in animal models of ALS…

33
…and in spinal cord of ALS patients FALS SOD1 A4V

34
Mutant SOD1 binds Bcl2 specifically in the mitochondria

35
SOD1 aggregates recruit Bcl2 and start apoptosis in FALS

36
Toxic mechanisms of SOD1 in mitochondria INSIDE THE MITOCHONDRIA *forming aggregates *interfering with cytochrome c in the peroxisomes (fusion of the peroxisomes membranes, formation of pores and release of cytochrome c, initiation of apoptosis) OUTSIDE THE MITOCHONDRIA *forming aggregates in the outer membrane, this leads to disruption of TOM (Translocator Outer Membrane complex)disruption of protein transport in the mitochondrion. *forming aggregates with anti-apoptotic Bcl2 at the surface of the mitochondria, thus leading to apoptosis

37
The mitochondrion as a target of mutant SOD1

38
How could mutant SOD1 be related to mitochondrial defects? Altering mitochondrial structure, causing formation of vacuoles and ultimately rupture of the mitochondrial outer membrane leading to apoptosis

39
Evidences that SOD1 could be involved also in SALS -Symptoms and pathology of SALS patients are the same as in SOD1 related FALS patients -Pathologic alterations of SOD1 mutant mice are similar to those observed in SALS patients Formation of mitochondrial vacuoles Expansion and rupture of the mitochondrial outer membrane Alterations of calcium homeostasis in the mitochondria Alterations of mitochondrial membrane potential Formation of calcium deposits within the mitochondria

40
1-SOD1 could be modified in SALS 2-SALS and FALS may share the same toxic mechanism of toxicity of SOD1

41
Can SOD1 form aggregates also in SALS?

43
wtSOD1 can be oxidized

44
SOD1 conformation-specific antibody

45
Conformation-specific SOD1 antibody detects oxSOD1 in non-denaturing conditions…

46
…but not in denaturing conditions: SOD1 oxidation as a mechanism to form SOD1 aggregates

47
SOD1 oxidation inhibits anterograde Fast Axonal Transport FAT

48
Sequestration of conformation-specific oxSOD1 reverts the effects of wtSOD1 on FAT in SALS

49
Conformation-specific SOD1 antibody reacts with wtSOD1 only in SALS

50
Mitochondrial abnormalities and dysfunction both in FALS and SALS A cause or a consequence?

51
SOD1 can be hyper-oxidized in certain forms of SALS

52
Hyper-oxidized SOD1 forms aggregates

53
Hyper-oxidized SOD1 forms a complex with Bcl2…

54
…and co-localizes with Bcl2

55
Hyper-oxidized SOD1 can be toxic to the mitochondria only by binding to Bcl2

56
Bcl2 is comformationally modified in lymphoblasts of hyper-oxidized SOD1 ALS and in fALS

57
Hyper-oxidized SOD1 can be associated with mitochondrial dysfunction

58
Defective axonal transport in ALS

59
Characteristic of ALS is axonal swelling www.afip.org

60
Axonal transport: Intracellular transport in neurons -Neurons are polarized cells. The presence of a positive or negative pole provides driving force to regulate anterograde or retrograde transport. -Anterograde: from cell body to neuronal end. Positive pole. -Retrograde: from neuronal end back to cell body. Negative pole. What is transported? Proteins (synthesized in the cell body) Organelles-Mitochondria Vesicles
 Organelles-Mitochondria Vesicles.")
61
Axonal Transport

62
Proteins regulating axonal tranport Kinesin 1 Anterograde transport Dynein/dynactin Retrograde transport

63
Point mutations in the gene of dynactin are associated to FALS

64
ALS and axonal transport Deposition of neurofilament Defects and mutations in dynein In motorneurons, axons are very long thus both anterograde and retrograde transport are complex events Neuronal transport is reduced in ALS SOD1 aggregates

65
Deposition of fibrillar proteinacious material in Amyotrophic Lateral Sclerosis (ALS) Ross and Poirier, 2004
 Ross and Poirier, 2004")
66
Axonal transport in ALS -Increased anterograde and decreased retrograde axonal transport in ALS patients. -Dynactin mutations associated with impaired retrograde transport -Decreased transport of mitochondria also in certain SOD1 mutants

67
Mechanisms of axonal transport defects: damage to mitochondria

68
Axonal vacuolation is an early event that precedes neurodegeneration in a model of ALS

69
Neuroreport Aggregates of G93ASOD1 are found in the axons, co-localizing with dynein: possible role of mutant SOD1 in damaging axonal transport

70
Model of aberrant interaction between SOD1 aggregates and dynein on FALS

71
SOD1 mutation in ALS: gain of toxic function rather than loss of physiologic function

72
SOD1siRNA improves ALS symptoms and viability in a SOD1G93A mutant mouse.

Similar presentations

>")
 (MND) are a group of neurological disorders that selectively affect motor neurons.>")



>")



 and the Quad SOD mutant: Implications for Amyotrophic Lateral Sclerosis Jesse Fitzpatrick.>")



>")




>")
