Download presentation
Presentation is loading. Please wait.

1
Chapter 3. Light emitting diod

2
Electron configuration

3
Covalent bond

4
Organic semiconductors:
extensively conjugated molecular systems such as fully conjugated polymers or cata-condensed polyacenes (PAH, e.g., anthracene)
")
6
p-electron clound in anthracene
Electron density sections through central molecular plane, with each contour line representing an increase of 500 electrons/nm3

7
Molecular orbitals

8
types of electronic transitions in polyatomic molecules

11
The total spin quantum number S = Σ si
The multiplicities M = 2S + 1

12
Spin of electrons

13
Statistical states Ms:Magnetic quantum number

14
Energetics of spin states
Externally applied H ~ 1 to 10 kG Energetics of spin states Zero-field splitting EZF ~ 0.1 cm1 ~ 1 kG Spin interaction Coulomb repulsion

15
Electronic excitation and de-excitation in organic molecules

16
Jabloński Diagram

19
probability of transition. The Beer-Lambert Law. Oscillator strength

20
The molecules whose absorption transition moments are parallel to the
electric vector of a linearly polarized incident light are preferentially excited. The probability of excitation is proportional to the square of the scalar product of the transition moment and the electric vector. This probability is thus maximum when the two vectors are parallel and zero when they are perpendicular.

21
Frank-Condon principle
Electronic transition ~ s Vibration ~ – s Vertical transition Bathochromic: red-shift Hyposochromic: blue-shift

22
Effects of domain overlap & corresponding wave amplitudes
Mirror image rule

24
Mirror image rule

25
Rigorous relatioship between absorption & fluorescence
Radiative & non-radiative processes Kasha’s rule

26
Internal conversion Fluorescence
Internal conversion is a non-radiative transition between two electronic states of the same spin multiplicity. From S1, internal conversion to S0 is possible but is less efficient than conversion from S2 to S1, because of the much larger energy gap between S1 and S0. Fluorescence Emission of photons accompanying the S1 S0 relaxation is called fluorescence. In most cases, the absorption spectrum partly overlaps the fluorescence spectrum. At r. t., a small fraction of molecules is in a vibrational level higher than 0 in the ground state as well as in the excited state. At low temp, this departure from the Stokes Law should disappear. It should be noted that emission of a photon is as fast as absorption of a photon (~1015 s). However, excited molecules stay in the S1 state for a certain time (a few tens of picoseconds to a few hundreds of nanoseconds, depending on the type of molecule and the medium) before emitting a photon or undergoing other de-excitation processes (internal conversion, intersystem crossing).
. However, excited molecules stay in the S1 state for a certain time (a few tens of picoseconds to a few hundreds of nanoseconds, depending on the type of molecule and the medium) before emitting a photon or undergoing other de-excitation processes (internal conversion, intersystem crossing).")
27
Experimental schematic for detection of the Stokes’ shift
The energy of the emission is typically less than that of absorption. Hence, fluorescence typically occurs at lower energies or longer wavelengths.

28
After the 1850s, Stokes became involved in administrative matters, and his scientific productivity decreased. Some things never change.

29
Stokes shift

30
Intersystem crossing and subsequent processes
is a non-radiative transition between two isoenergetic vibrational levels belonging to electronic states of different multiplicities. Delayed fluorescence T1S1 can occur when the energy difference between S1 & T1 is small and when the lifetime of T1 is long enough. Triplet-triplet annihilation In concentrated solutions, a collision between two molecules in the T1 state can provide enough energy to allow one of them to return to the S1 state leads to a delayed fluorescence emission. Triplet-triplet transitions Once a molecule is excited and reaches triplet state T1, it can absorb a different wavelength because triplet-triplet transitions are spin allowed.

31
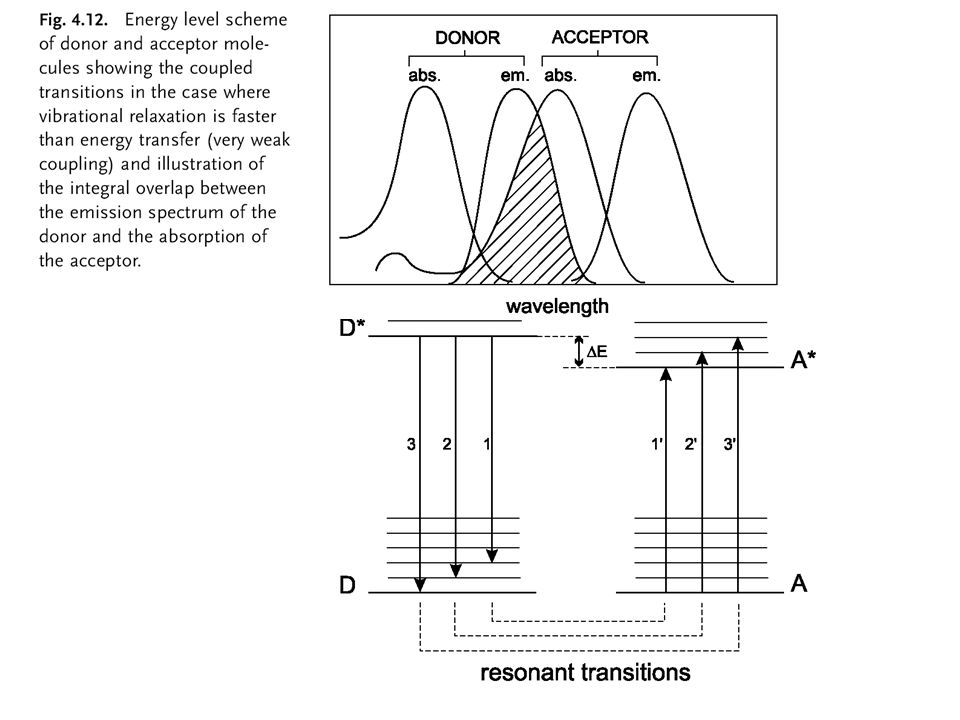
Energy transfer

33
Distinction between radiative and non-radiative transfer

34
hopping

35
Excitation energy transfer
heterotransfer homotransfer

38
Radiative energy transfer

39
(rate constant) (lifetime of excited state)
 (lifetime of excited state)")
40
Non-radiative energy transfer
EET: excitation energy transfer or electronic energy transfer RET: resonance energy transfer FRET: fluorescence resonance energy transfer

42
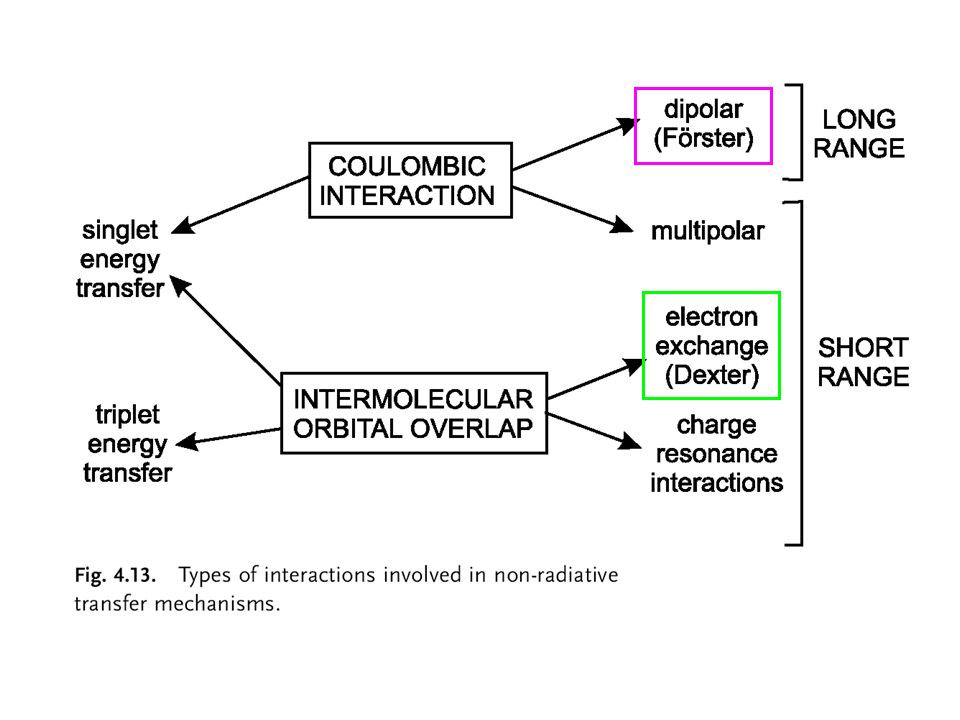
Energy transfer can result from different interaction mechanisms
Energy transfer can result from different interaction mechanisms. The interactions may be Coulombic and/or due to intermolecular orbital overlap. * * The Coulombic interactions consist of long-range dipole-dipole interactions (Forster’s mechanism) and short-range multi-polar interactions. * * The interactions due to intermolecular orbital overlap, which include electron exchange (Dexter’s mechanism) and charge resonance interactions, are of course only short range. It should be noted that for singlet-singlet energy transfer, all types of interactions are involved, whereas triplet-triplet energy transfer is due only to orbital overlap.
 and short-range multi-polar interactions. * * The interactions due to intermolecular orbital overlap, which include electron exchange (Dexter’s mechanism) and charge resonance interactions, are of course only short range. It should be noted that for singlet-singlet energy transfer, all types of interactions are involved, whereas triplet-triplet energy transfer is due only to orbital overlap.")
44
CI: Coulombic interaction
EE: electron exchange The total interaction energy can be expressed as a sum of two terms: a Columbic term Uc and an exchange term Uex. The Coulombic term corresponds to the energy transfer process in which the initially excited electron on the donor D returns to the ground state orbital on D, while simultaneously an electron on the acceptor A is promoted to the excited state. The exchange term corresponds to an energy transfer process associated with an exchange of two electrons between D and A.

45
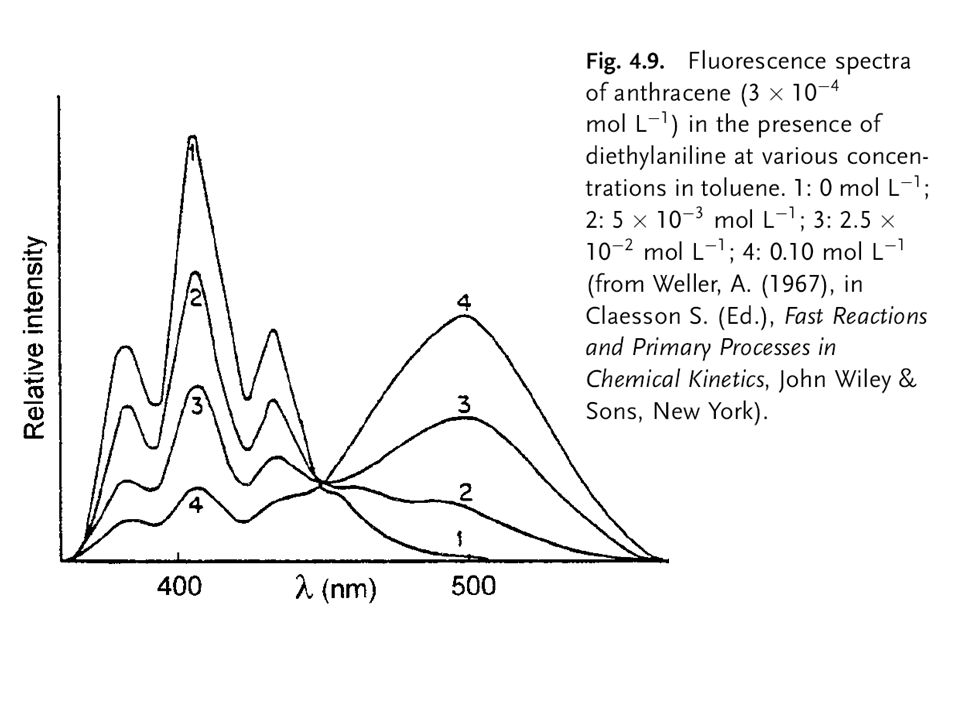
Formation of excimers and exciplexes

46
Excimers An excimer is located at wavelengths higher than that of the monomer and does not show vibronic bands.

48
P3HT PL heating-1

50
Exciplexes

52
Lifetimes and quantum yields
Excited-state lifetimes The rate constants for the various processes will be denoted as follows:

54
A very short pulse of light at time 0 brings a certain number of molecules A to the S1 excited state by absorption of photons. These excited molecules then return to So. The rate of disappearance of excited molecules is expressed by the following differential equation:

55
iF(t), the d-pulse response of the system, decrease according to a single expontial.
, the d-pulse response of the system, decrease according to a single expontial.")
56
Quantum yields

57
τr = 1/krs When the fluorescence quantum yield and the excited-state lifetime of a fluorophore are measured under the same conditions, the non-radiative and radivative rate can be easily calculated by means of the following relations:

59
quenching The sum kM of the rate constants for de-excitation processes is equal to the reciprocal excited-state lifetime t0: Kq : the observed rate constant for the bimolecular process

60
The fluorescence characteristics (decay time and/or fluorescence quantum yield) of M* are affected by the presence of Q as a result of competition between the intrinsic de-excitation and these intermolecular processes.
 of M* are affected by the presence of Q as a result of competition between the intrinsic de-excitation and these intermolecular processes.")
62
Overview of the intermolecular de-excitation of excited molecules leading to fluorescence quenching
4.2.1 Phenomenological approach (static quenching)
")
63
Dynamic quenching Stern-Volmer kinetics (ignore the transient effects)
")
64
i(t) = i(0) exp (-t/t)
 = i(0) exp (-t/t)")
65
KSV : Stern-Volmer constant
I0/I vs. [Q] Stern-Volmer plot

66
Static quenching The term static quenching implies either the existence of a sphere of effective quenching or the formation of a ground-state non-fluorescent complex (Figure 4.1)
")
67
Sphere of effective quenching

70
Formation of a ground-state non-fluorescent complex

71
I ~ [M]
![I ~ [M]](http://slideplayer.com/slide/8555906/26/images/71/I+%EF%BD%9E+%5BM%5D.jpg "I ~ [M]")
72
Simultaneous dynamic and static quenching
non-fluorescent complex

73
static Sphere of effective quenching

Similar presentations

















, but how to study in cells? Do rafts really exist in cells? Are.>")









 Laser Flash Photolysis This.>")



 Quantum-Mechanical Properties Of Light Photoelectric Effect Photoelectric Effect Energy States of.>")



