Download presentation
Presentation is loading. Please wait.

1
Sequencing and Assembly GEN875, Genomics and Proteomics, Fall 2010

2
History of DNA Sequencing Avery: Proposes DNA as ‘Genetic Material’ Watson & Crick: Double Helix Structure of DNA Holley: Sequences Yeast tRNA Ala 1870 1953 1940 1965 1970 1977 1980 1990 2002 Miescher: Discovers DNA Wu: Sequences Cohesive End DNA Sanger: Dideoxy Chain Termination Gilbert: Chemical Degradation Messing: M13 Cloning Hood et al.: Partial Automation Cycle Sequencing Improved Sequencing Enzymes Improved Fluorescent Detection Schemes 1986 Next Generation Sequencing Improved enzymes and chemistry Improved image processing Adapted from Francis Oulette; Adapted from Eric Green, NIH; Adapted from Messing & Llaca, PNAS (1998) 1 15 150 50,000 25,000 1,500 200,000 50,000,000 Efficiency (bp/person/year) 15,000 100,000,000,000 2008
 ,000 25,000 1, ,000 50,000,000 Efficiency (bp/person/year) 15, ,000,000,")
3
Explosive Growth in Sequencing 8/22/2005 Press Release: INSD (GenBank, EMBL, DDBJ) reaches 100 Gigabase milestone
 reaches 100 Gigabase milestone")
4
What do we sequence? Genomes (de novo, resequencing) Metagenomes or complex samples Transcripts Fragments recovered by chIP or tagged in some other way
 Metagenomes or complex samples Transcripts Fragments recovered by chIP or tagged in some other way.")
5
NCBI Genomes http://www.ncbi.nlm.nih.gov/Genomes/ Eukaryotic Genomes: Complete 23, 25, 22, 20 Assembly 230, 162, 109, 72 In progress 229, 235, 299, 166 Prokaryotic Genomes: Complete 745, 567, 371, 254 In progress 1215, 841, 615, 433 Comparison of data from 9/4/08, 9/5/07, 9/4/06 and 8/31/05

6
NCBI Genomes 9/6/2010

8
Sequencing Platforms Sanger sequencing and capillary electrophoresis Massively parallel pyrosequencing (454) “proprietary Clonal Single Molecule Array technology and novel reversible terminator- based sequencing” (Illumina) Sequencing by ligation (ABI SOLiD) Single molecule sequencing (PacBio)
 proprietary Clonal Single Molecule Array technology and novel reversible terminator- based sequencing (Illumina) Sequencing by ligation (ABI SOLiD) Single molecule sequencing (PacBio)")
9
Basics of the “old” technology Clone the DNA. Generate a ladder of labeled (colored) molecules that are different by 1 nucleotide. Separate mixture on some matrix. Detect fluorochrome by laser. Interpret peaks as string of DNA. Strings are 500 to 1,000 letters long 1 machine generates 57,000 nucleotides/run Assemble all strings into a genome. Adapted from Francis Oulette
 molecules that are different by 1 nucleotide. Separate mixture on some matrix. Detect fluorochrome by laser. Interpret peaks as string of DNA. Strings are 500 to 1,000 letters long 1 machine generates 57,000 nucleotides/run Assemble all strings into a genome. Adapted from Francis Oulette.")
10
Sample Isolate DNA Physical fragmentation Size selection Ligate randomly into vectors Transformation Plate on agar Pick and grow individual colonies Isolate cloned constructs Cycle Sequencing High-throughput Steps Library construction and sequencing

11
Dual Ended Sequencing Can Provide Information to Link Contigs 5 Kb insert Primer A Primer B Sequencing with primers that begin in the vector on either side of the insert yields about 800 bp of DNA sequence from each end of the insert The middle of the insert is never sequenced for most clones used in the project

12
Basics of the “new” technology Get DNA. Attach it to something. Extend and amplify signal with some labeling scheme. Detect fluorochrome by microscopy. Interpret series of spots as short strings of DNA. Strings are 30-300 letters long Multiple images are interpreted as 0.4 to 1.2 GB/run (1,200,000,000 letters/day). Map or align strings to one or many genome or assemble. Adapted from Francis Oulette
. Map or align strings to one or many genome or assemble. Adapted from Francis Oulette.")
13
Differences between the various platforms: Nanotechnology used. Resolution of the image analysis. Chemistry and enzymology. Signal to noise detection in the software Software/images/file size/pipeline Cost $$$ Adapted from Francis Oulette

14
Next Generation DNA Sequencing Technologies Adapted from Richard Wilson, School of Medicine, Washington University, “Sequencing the Cancer Genome” http://tinyurl.com/5f3alk 3 Gb ==

15
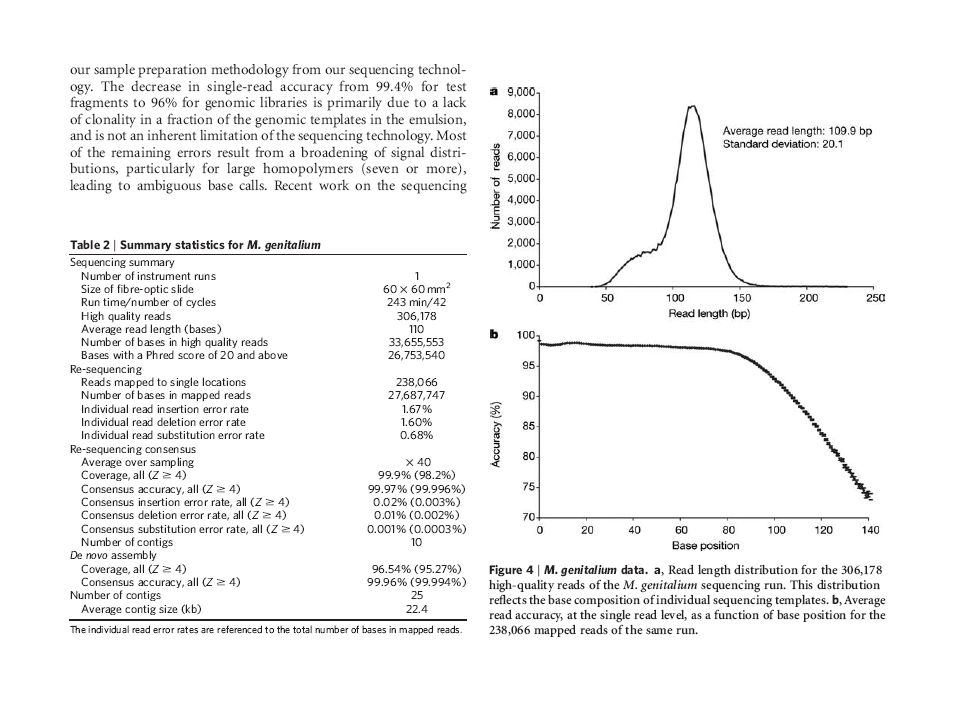
Roche 454 Genome sequencing in microfabricated high-density picolitre reactors Margulies, M. Eghold, M. et al. Nature. 2005 Sep 15; 437(7057):326-7 Pyrosequencing
:326-7 Pyrosequencing.")
17
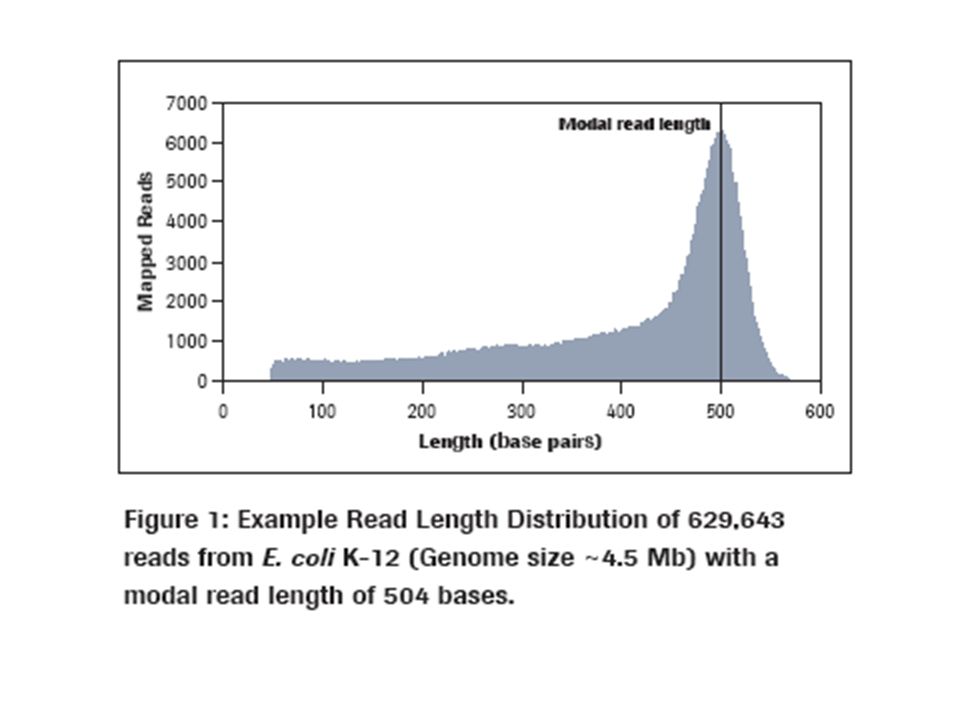
Throughput 400-600 million high-quality, filter-passed bases per run* 1 billion bases per day Run Time10 hours Read LengthAverage length = 400 bases Accuracy Q20 read length of 400 bases (99% at 400 bases and higher for prior bases) Reads per run>1 million high-quality reads GS FLX Titanium Series: The GS FLX Titanium series reagents run on the Genome Sequencer FLX Instrument, a system based on 454's sequencing-by-synthesis technology. The GS FLX Titanium series improves on the current system with upgraded reagents, consumables, and software. sequencing-by-synthesis technology GS FLX Titanium Series:

19
Solexa-based Whole Genome Sequencing Adapted from Richard Wilson, School of Medicine, Washington University, “Sequencing the Cancer Genome” http://tinyurl.com/5f3alk

20
Solexa-based Whole Genome Sequencing Solexa flow cell ~50M clusters are sequenced per flow cell. Adapted from Richard Wilson, School of Medicine, Washington University, “Sequencing the Cancer Genome” http://tinyurl.com/5f3alk

21
Debbie Nickerson, Department of Genome Sciences, University of Washington, http://tinyurl.com/6zbzh4

23
Genome-scale Sequence Analysis De novo assembly Templated assembly Read mapping or alignment to a reference genome

24
“The choice of alignment or assembly algorithm is strongly influenced by both the experiment in question and the details of the sequencing technology used. The performance characteristics of the sequencing machines are changing rapidly, and any delineation of performance characteristics such as machine capacity, run time or read length and its relationship to error profile will quickly be outdated.”

25
Assemblers Greedy Assemblers – compare all reads to each other then join them in order of overlap size Figure 8. Greedy assembly of four reads.

26
Assemblers Overlap Graph Assemblers – make a graph where each node represents a read and edges between them represent overlaps. Figure 9. Overlap graph for a bacterial genome. The thick edges in the picture on the left (a Hamiltonian cycle) correspond to the correct layout of the reads along the genome (figure on the right). The remaining edges represent false overlaps induced by repeats (exemplified by the red lines in the figure on the right)
 correspond to the correct layout of the reads along the genome (figure on the right). The remaining edges represent false overlaps induced by repeats (exemplified by the red lines in the figure on the right).")
27
Assembly with dual-ended sequencing Sequence assembly Contigs linked by a spanning clone Contigs joined by overlaps Scaffold – two or more linked contigs

28
Repeat handling Screen out known repeats and set them aside for later Infer repetitiveness based on coverage First assemble unambiguous overlaps, then resolve repeats using mate pairs

29
Assemblers and short reads Full overlap assemblers compare all reads against all other reads. Scale quadratically with the number of reads. Computationally intractable for large NSG datasets Led to development of k-mer based methods: a de Bruijn graph with a node for every k-mer observed in the sequence set and an edge between nodes if these two k-mers are observed adjacently in a read

30
Figure 2. Differences between an overlap graph and a de Bruijn graph for assembly. Based on the set of 10 8-bp reads (A), we can build an overlap graph (B) in which each read is a node, and overlaps >5 bp are indicated by directed edges. Transitive overlaps, which are implied by other longer overlaps, are shown as dotted edges. In a de Bruin graph (C ), a node is created for every k-mer in all the reads; here the k-mer size is 3. Edges are drawn between every pair of successive k-mers in a read, where the k-mers overlap by k 1 bases. In both approaches, repeat sequences create a fork in the graph. Note here we have only considered the forward orientation of each sequence to simplify the figure.
, we can build an overlap graph (B) in which each read is a node, and overlaps >5 bp are indicated by directed edges. Transitive overlaps, which are implied by other longer overlaps, are shown as dotted edges. In a de Bruin graph (C ), a node is created for every k-mer in all the reads; here the k-mer size is 3. Edges are drawn between every pair of successive k-mers in a read, where the k-mers overlap by k 1 bases. In both approaches, repeat sequences create a fork in the graph. Note here we have only considered the forward orientation of each sequence to simplify the figure..")
31
Figure 1. The k-mer uniqueness ratio for five well-known organisms and one single-celled human parasite. The ratio is defined here as the percentage of the genome that is covered by unique sequences of length k or longer. The horizontal axis shows the length in base pairs of the sequences. For example, ;92.5% of the grapevine genome is contained in unique sequences of 100 bp or longer.

32
De Bruijn k-mer assemblers Newbler (Roche 454) SHARCGS VCAKE VELVET EULER-SR EDENA ABySS ALLPATHS SOAPdenovo Contrail
 SHARCGS VCAKE VELVET EULER-SR EDENA ABySS ALLPATHS SOAPdenovo Contrail")
33
Most assemblers have an error detection and resolution phase Errors produce characteristic graphic structures

34
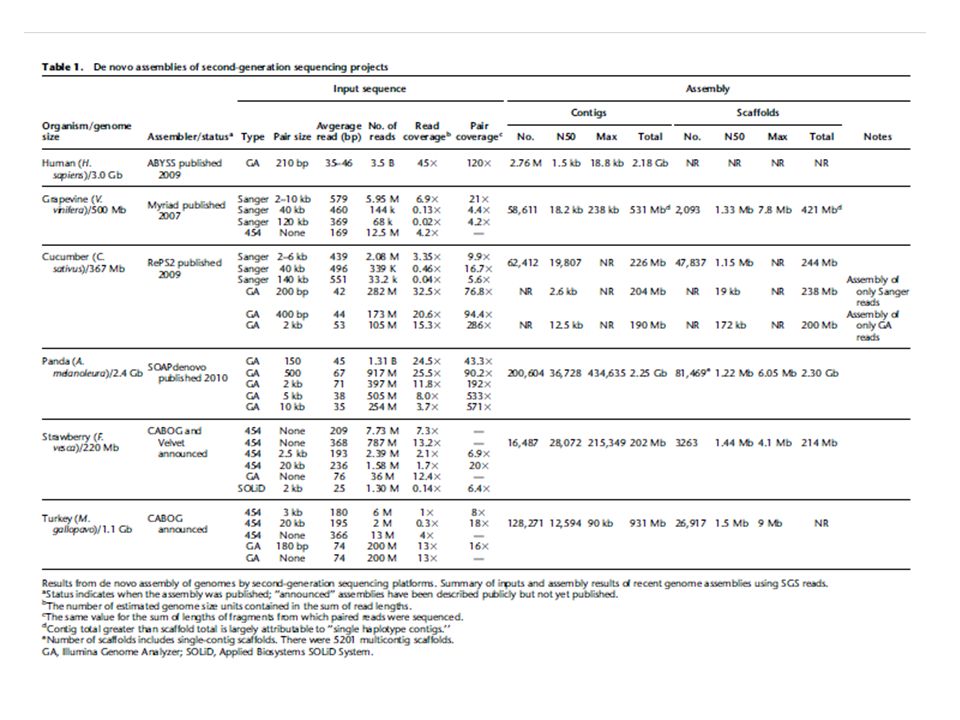
Problems with de Bruijn graph methods Require large amount of memory to store graph – for example Velvet would require a terabyte of memory to assemble the human genome Not as easy to parallelize as overlap assemblers From Shatz et al. 2010: “To date, only two de Bruijn graph assemblers have been shown to have the ability to assemble a mammalian-sized genome. ABySS (Simpson et al. 2009) assembled a human genome in 87 h on a cluster of 21 eight-core machines each with 16 GB of RAM (168 cores, 336 GB of RAM total). SOAPdenovo assembled a human genome in 40 h using a single computer with 32 cores and 512 GB of RAM (Li et al. 2010). Although these types of computing resources are not widely available, they are within reach for large-scale scientific centers.”
 assembled a human genome in 87 h on a cluster of 21 eight-core machines each with 16 GB of RAM (168 cores, 336 GB of RAM total). SOAPdenovo assembled a human genome in 40 h using a single computer with 32 cores and 512 GB of RAM (Li et al. 2010). Although these types of computing resources are not widely available, they are within reach for large-scale scientific centers. .")
36
How many clones/reads do we need? …according to the work of Lander and Waterman (Genomic mapping by fingerprinting random clones: a mathematical analysis.Genomics. 1988 Apr;2(3):231-9.), the number of “islands” or contigs formed from randomly collected sequences depends on: G= Genome Length L = Sequence Read Length N = Number of Sequences Collected T= Number of Basepairs of Overlap Needed # Islands =Ne LN G TLTL (- ( 1 - )
:231-9.), the number of islands or contigs formed from randomly collected sequences depends on: G= Genome Length L = Sequence Read Length N = Number of Sequences Collected T= Number of Basepairs of Overlap Needed # Islands =Ne LN G TLTL (- ( 1 - ).")
37
5 Mbp Genome, 500 bp reads, 25 bp overlap # readscoverage% sequenced# contigs 25000.2522.121971 50000.539.353109 10000163.213867 20000286.472991 30000395.021735 40000498.17895 50000599.33433 60000699.75201 70000799.9191 80000899.9740 90000999.9917 10000010100.007

38
Graph of previous data

39
Shotgun Sequencing Model 0 500 1000 1500 2000 2500 010,000 20,000 30,00040,00050,00060,00070,000 # Sequences # non-singleton contigs Predicted 5.5 Mb size Observed # non-singletons Predicted 3.7Mb size Genome size as predicted from the assembly

40
Figure 3. Expected average contig length for a range of different read lengths and coverage values. Also shown are the average contig lengths and N50 lengths for the dog genome, assembled with 710-bp reads, and the panda genome, assembled with reads averaging 52 bp in length.

41
Combining sequence data types In practice, appears to be the best strategy for both microbial and eukaryotic genomes Creates assembly challenges of its own

42
One strategy for microbial genomes ~¼ run of 454 regular, ¼ run of paired end (2.5 kb library) plus one lane of Solexa Assemble Solexa data with Velvet Assemble 454 data with Newbler Shred the Velvet assembly into Newbler size reads and add it to the 454 assembly Use Solexa deep coverage to “polish”
 plus one lane of Solexa Assemble Solexa data with Velvet Assemble 454 data with Newbler Shred the Velvet assembly into Newbler size reads and add it to the 454 assembly Use Solexa deep coverage to polish")
44
Gap Closure Strategies Primer walk to sequence the rest of linking clones that span a scaffold gap Primer walk off clones at the ends of contigs for which there is no linking information PCR based on your best guess at contig order (comparison to other closely related genomes, predicted genes at the end of genomes, anything else you can come up with) Combinatorial PCR with primers designed at the end of each contig
 Combinatorial PCR with primers designed at the end of each contig")
45
Phred Scores Phred Score P( incorrect base )Base call accuracy 10 1 in 10 90% 20 1 in 100 99% 30 1 in 1000 99.9% 40 1 in 10000 99.99% 50 1 in 100000 99.999%
Base call accuracy 10 1 in 10 90% 20 1 in % 30 1 in % 40 1 in % 50 1 in %")
Similar presentations




 Isolate total RNA Isolate mRNA from total RNA (poly.>")








 National University of Sciences and Technology.>")




 FISH 2) “Restriction mapping” 3) Southern analysis : DNA 4) Northern analysis: RNA tells size tells which tissues or conditions.>")
