Download presentation
Presentation is loading. Please wait.

1
Error model for massively parallel (454) DNA sequencing Sriram Raghuraman (working with Haixu Tang and Justin Choi)
 DNA sequencing Sriram Raghuraman (working with Haixu Tang and Justin Choi)")
2
Sequencing Preparation Randomly fragment entire genome Nebulize fragments. Add adapters. Attach to DNA capture beads in water oil emulsion PCR amplify fragments attached to beads Place beads bound to multiple copies of same fragment in a PicoTiterPlate. Add enzymes including polymerase and luciferase.

3
Sequencing Process Place plates in a sequencer. Wash nucleotides (A,C,G,T) in series over plate. When a complementary nucleotide enters a well, the template strand is extended by DNA polymerase. Addition of the nucleotide releases light which is recorded by a CCD camera. Hundreds of thousands of beads are then sequenced in parallel. Genome sequencing in microfabricated high-density picolitre reactors-Nature 437, 376-380 (15 September 2005)
.")
4
Speed of sequencing ~25 million bases at >=99% accuracy in a 4 hour run ~230,000 reads Average read length 110 bases

5
Data Sets(Newbler) 984766 reads aligned by Newbler Bases98878209 Matches97793963 (98.90%) Mismatches10643(0.01%) Inserts368332(0.37%) Deletes668451 (0.67%) ‘N’ terms36820(0.03%)
 reads aligned by Newbler Bases Matches (98.90%) Mismatches10643(0.01%) Inserts368332(0.37%) Deletes (0.67%) ‘N’ terms36820(0.03%)")
6
Data Set (Sanger) Staphylococcus aureus subsp. aureus COL from NCBI Assembly Archive 50000 reads Bases27173366 Matches27094113(99.70%) Mismatches71203(0.26%) Inserts1827(0.006%) Deletes6223(0.02%)
 Mismatches71203(0.26%) Inserts1827(0.006%) Deletes6223(0.02%).")
7
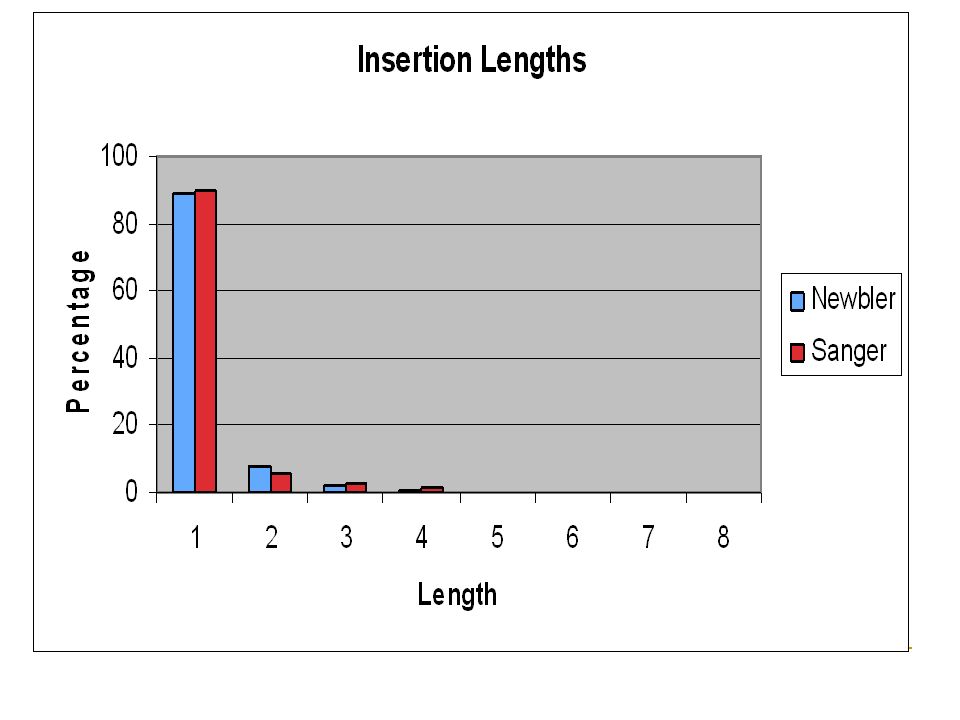
Length Distributions Newbler reads are shorter than Sanger reads Newbler Average read length ~100 bases Sanger Average read length ~545 bases

10
Accuracy % Newbler reads show a prevalence of gaps as compared to mismatches Newbler mismatches are indirect AA-CT AAG-T Sanger reads contain more mismatches than gaps

11
Biases in Substitutions and Gaps

12
Substitutions

13
The case for homogeneous gaps

14
Homogeneous gaps Newbler reads often exhibit homogeneous gaps Insertions R:-CGGGATCAGTGATGGCGTACGTTTACCGGGTTAAAAGAGGGCCGG G:-CGGGATCAGTGATG-CG-A--TT--CCGG-TTAAA-GAGG-C-GG Deletions R:-TTTACA-TCGTGGTCGTGACAC-ATCGACACTGTAT-AAAA-CCAT G:-TTT-CAATC-TGGTCGTGACACCATCGACACTGTATTAAAAACCAT

15
Insert Transitions

16
Delete Transitions

17
Insert Strings

18
Delete Strings

19
Some examples Blast 1 st hit CTCCGCATC-AAAG....TTT-GATGCGGAG CTCCGCATCCAAAG....TTTGGATGCGGAG Newbler Alignment CCTCCGCATC-AAAG....TTTG-ATGCGGAG C-TCCGCATCCAAAG....TTTGGATGCGGAG No difference between homogeneous and regular gaps as far as BLAST is concerned

20
Markov Model

21
General Ideas Incorporate provisions for homogeneous gaps Train model on Newbler data A Markov model that accounts for homogeneous gaps should perform better than one that doesn’t (i.e. BLAST)
.")
22
MM AA MM-MisMatch CCGGTTA-C-G-T--A-C-G-T AC AG AT

23
Procedure Get initial, transition and emission probabilities from Newbler reads Use Markov model to perform pairwise alignment of unaligned reads by employing Viterbi’s algorithm Compare results to BLAST alignment of same reads

24
Procedure Get initial, transition and emission probabilities from Newbler reads Use Markov model to perform pairwise alignment of unaligned reads by employing Viterbi’s algorithm Compare results to BLAST alignment of same reads

25
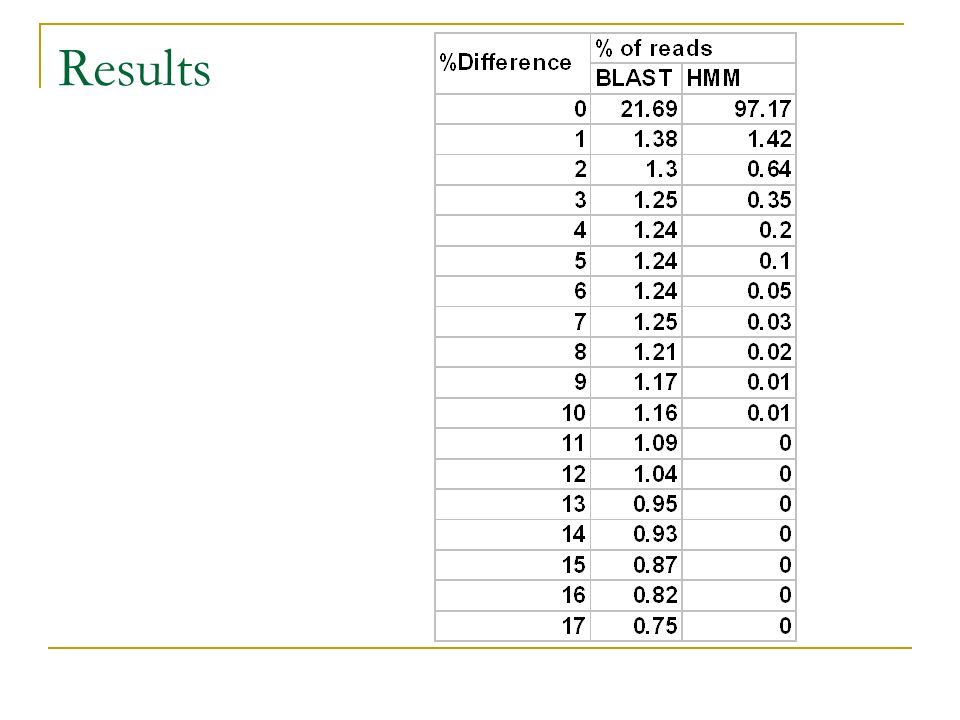
Results

27
Limitations Global Alignment only Local Alignment hinges on good alignment extension metric/method

Similar presentations



 Two strands of DNA run antiparallel.>")


 Isolate total RNA Isolate mRNA from total RNA (poly.>")





 Isolate total RNA Isolate mRNA from total RNA (poly.>")







