Download presentation
Presentation is loading. Please wait.

1
Solid State Computing Peter Ballo

2
Models Classical: Quantum mechanical: H = E Semi-empirical methods Ab-initio methods

3
Molecular Mechanics atoms = spheres bonds = springs math of spring deformation describes bond stretching, bending, twisting Energy = E(str) + E(bend) + E(tor) + E(NBI)
 + E(bend) + E(tor) + E(NBI)")
4
From ab initio to (semi) empirical Quantum calculation First principles Reliability proven within the approximations Basis sets, functional, all-electron or pseudo- potential.. Computationally expensive Based on fitting parameters Two body, three body…, multi-body potential No theoretical background empirical Applicability to large system no self consistency loop and no eigenvalue computation Reliability ?

5
DFT: the theory Schroedinger’s equation Hohenberg-Kohn Theorem Kohn-Sham Theorem Simplifying Schroedinger’s LDA, GGA Elements of Solid State Physics Reciprocal space Band structure Plane waves And then ? Forces (Hellmann-Feynman theorem) E.O., M.D., M.C. … The Framework of DFT
 E.O., M.D., M.C. … The Framework of DFT.")
6
Using DFT Practical Issues Input File(s) Output files Configuration K-points mesh Pseudopotentials Control Parameters LDA/GGA ‘Diagonalisation’ Applications Isolated molecule Bulk Surface
 Output files Configuration K-points mesh Pseudopotentials Control Parameters LDA/GGA ‘Diagonalisation’ Applications Isolated molecule Bulk Surface")
7
The Basic Problem Dangerously classical representation Cores Electrons

8
Schroedinger’s Equation Hamiltonian operator Kinetic Energy Potential Energy Coulombic interaction External Fields Very Complex many body Problem !! (Because everything interacts) Wave function Energy levels
 Wave function Energy levels.")
9
First approximations Adiabatic (or Born-Openheimer) Electrons are much lighter, and faster Decoupling in the wave function Nuclei are treated classically They go in the external potential
 Electrons are much lighter, and faster Decoupling in the wave function Nuclei are treated classically They go in the external potential")
10
Self consistent loop Solve the independents K.S. =>wave functions From density, work out Effective potential New density ‘=‘ input density ?? Deduce new density from w.f. Initial density Finita la musicaYES NO

11
DFT energy functional Exchange correlation funtional Contains: Exchange Correlation Interacting part of K.E. Electrons are fermions (antisymmetric wave function)
.")
12
Exchange correlation functional At this stage, the only thing we need is: Still a functional (way too many variables) #1 approximation, Local Density Approximation: Homogeneous electron gas Functional becomes function !! (see KS3) Very good parameterisation for Generalised Gradient Approximation: GGA LDA
 Very good parameterisation for Generalised Gradient Approximation: GGA LDA.")
13
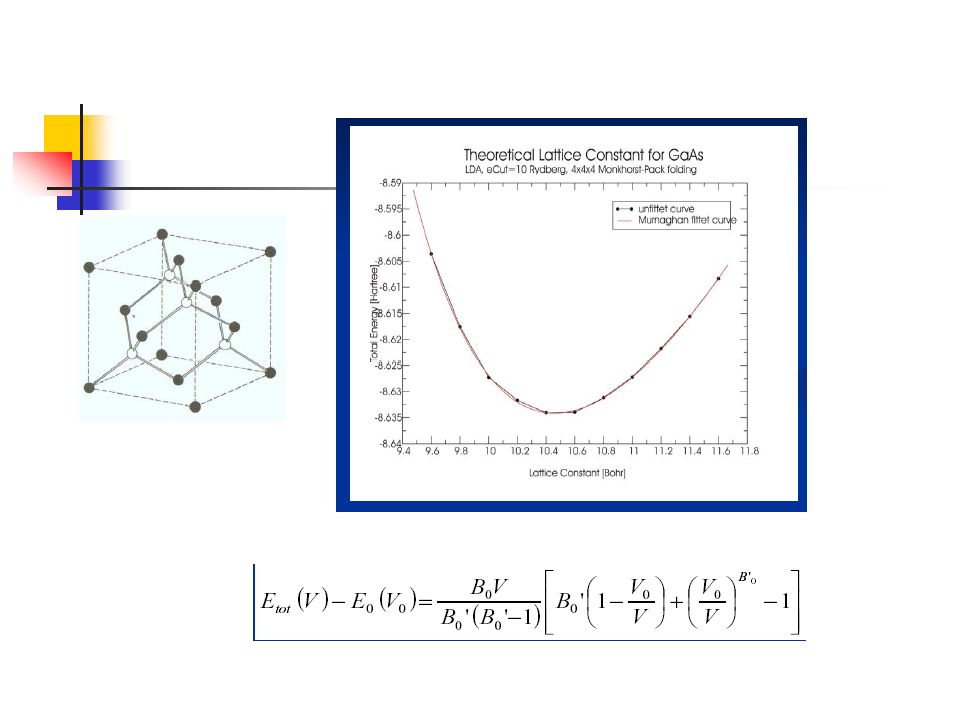
Bulk properties zero temperature equations of state (bulk modulus, lattice constant, cohesive energy) structural energy difference (FCC,HCP,BCC)
 structural energy difference (FCC,HCP,BCC)")
15
M. I. Baskes, Phys. Rev. B 46, 2727 (1992) M. I. Baskes, Matter. Chem. Phys. 50, 152 (1997)
 M. I. Baskes, Matter. Chem. Phys. 50, 152 (1997)")
16
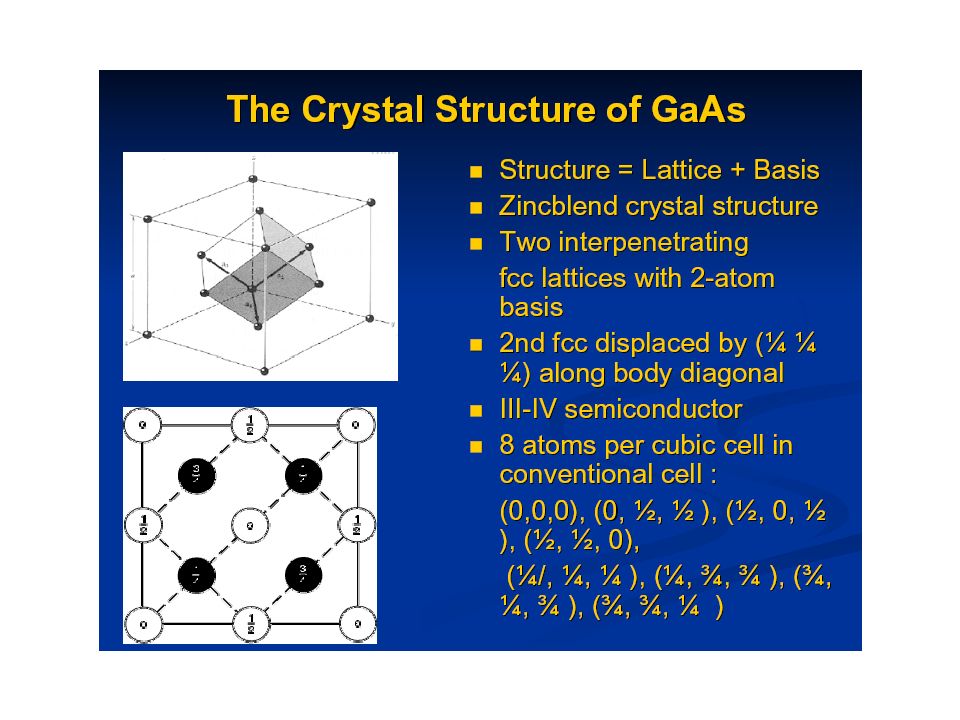
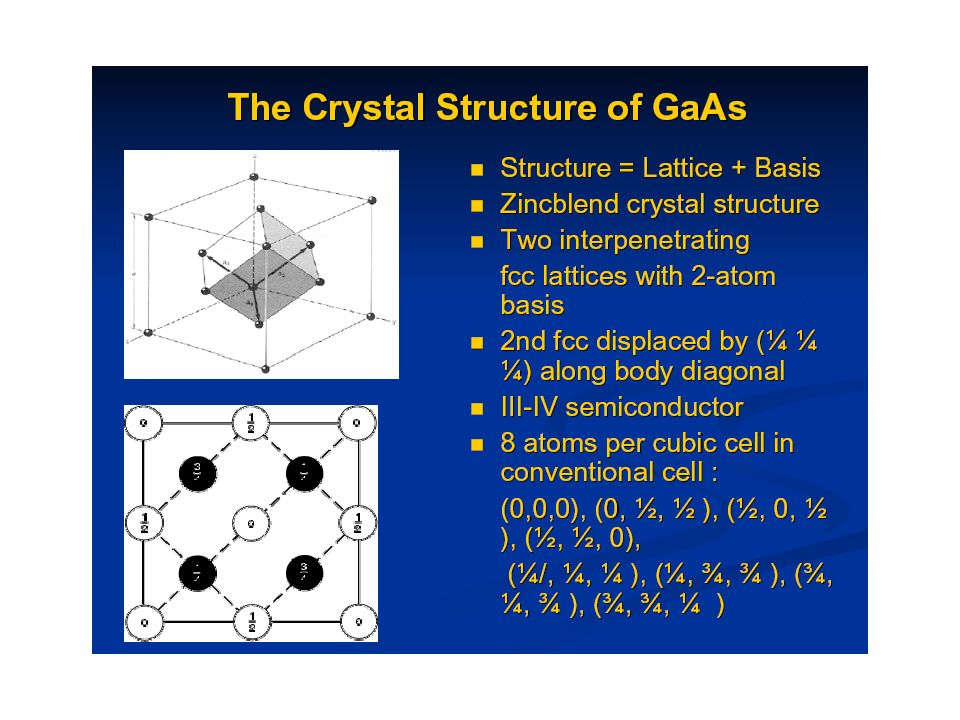
And now, for something completely different: A little bit of Solid State Physics Crystal structurePeriodicity

17
Reciprocal space Real Space a i Reciprocal Space b i Brillouin Zone (Inverting effect) k-vector (or k-point) sin(k.r) See X-Ray diffraction for instance Also, Fourier transform and Bloch theorem
 k-vector (or k-point) sin(k.r) See X-Ray diffraction for instance Also, Fourier transform and Bloch theorem")
18
Band structure Molecule E Crystal Energy levels (eigenvalues of SE)
")
19
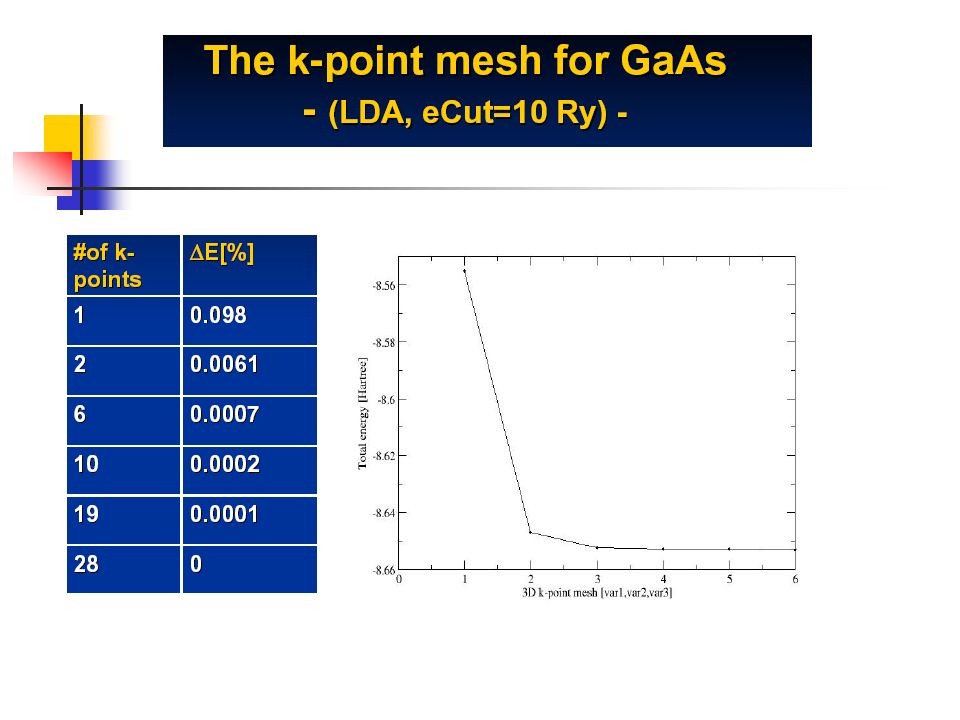
The k-point mesh Brillouin Zone (6x6) mesh Corresponds to a supercell 36 time bigger than the primitive cell Question: Which require a finer mesh, Metals or Insulators ??
 mesh Corresponds to a supercell 36 time bigger than the primitive cell Question: Which require a finer mesh, Metals or Insulators")
20
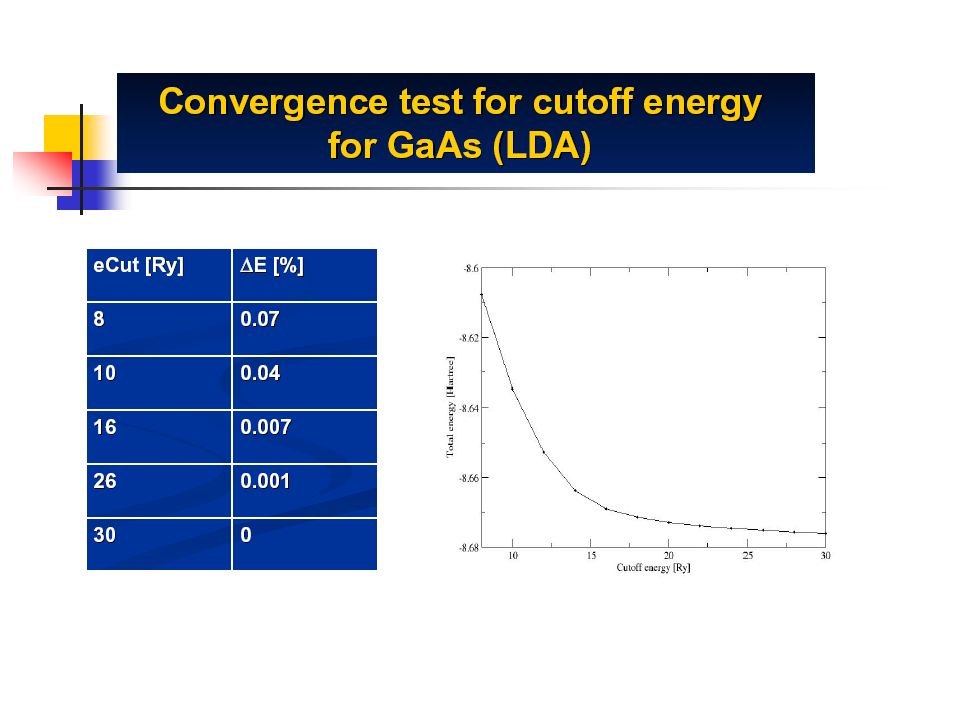
Plane waves Project the wave functions on a basis set Tricky integrals become linear algebra Plane Wave for Solid State Could be localised (ex: Gaussians) ++= Sum of plane waves of increasing frequency (or energy) One has to stop: E cut
 ++= Sum of plane waves of increasing frequency (or energy) One has to stop: E cut")
21
Solid State: Summary Quantities can be calculated in the direct or reciprocal space k-point Mesh Plane wave basis set, E cut

23
if (i.LE.n) then kx=kx-step ! Move to the Gamma point (0,0,0) ky=ky-step kz=kz-step xk=xk+step else if ((i.GT.n).AND.(i.LT.2*n)) then kx=kx+2.0*step ! Now go to the X point (1,0,0) ky=0.0 kz=0.0 xk=xk+step else if (i.EQ.2*n) then kx=1.0 ! Jump to the U,K point ky=1.0 kz=0.0 xk=xk+step else if (i.GT.2*n) then kx=kx-2.0*step ! Now go back to Gamma ky=ky-2.0*step kz=0.0 xk=xk+step end if
 ky=ky-step kz=kz-step xk=xk+step else if ((i.GT.n).AND.(i.LT.2*n)) then kx=kx+2.0*step . Now go to the X point (1,0,0) ky=0.0 kz=0.0 xk=xk+step else if (i.EQ.2*n) then kx=1.0 . Jump to the U,K point ky=1.0 kz=0.0 xk=xk+step else if (i.GT.2*n) then kx=kx-2.0*step . Now go back to Gamma ky=ky-2.0*step kz=0.0 xk=xk+step end if.")
29
# Crystalline silicon : computation of the total energy # #Definition of the unit cell acell 3*10.18 # This is equivalent to 10.18 10.18 10.18 rprim 0.0 0.5 0.5 # In lessons 1 and 2, these primitive vectors 0.5 0.0 0.5 # (to be scaled by acell) were 1 0 0 0 1 0 0 0 1 0.5 0.5 0.0 # that is, the default. #Definition of the atom types ntypat 1 # There is only one type of atom znucl 14 # The keyword "znucl" refers to the atomic number of the # possible type(s) of atom. The pseudopotential(s) # mentioned in the "files" file must correspond # to the type(s) of atom. Here, the only type is Silicon. #Definition of the atoms natom 2 # There are two atoms typat 1 1 # They both are of type 1, that is, Silicon. xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom 0.0 0.0 0.0 # Triplet giving the REDUCED coordinate of atom 1. 1/4 1/4 1/4 # Triplet giving the REDUCED coordinate of atom 2. # Note the use of fractions (remember the limited # interpreter capabilities of ABINIT)
 of atom. The pseudopotential(s) # mentioned in the files file must correspond # to the type(s) of atom. Here, the only type is Silicon. #Definition of the atoms natom 2 # There are two atoms typat 1 1 # They both are of type 1, that is, Silicon. xred # This keyword indicate that the location of the atoms # will follow, one triplet of number for each atom # Triplet giving the REDUCED coordinate of atom 1. 1/4 1/4 1/4 # Triplet giving the REDUCED coordinate of atom 2. # Note the use of fractions (remember the limited # interpreter capabilities of ABINIT).")
30
#Definition of the planewave basis set ecut 8.0 # Maximal kinetic energy cut-off, in Hartree #Definition of the k-point grid kptopt 1 # Option for the automatic generation of k points, taking # into account the symmetry ngkpt 2 2 2 # This is a 2x2x2 grid based on the primitive vectors nshiftk 4 # of the reciprocal space (that form a BCC lattice !), # repeated four times, with different shifts : shiftk 0.5 0.5 0.5 0.5 0.0 0.0 0.0 0.5 0.0 0.0 0.0 0.5 # In cartesian coordinates, this grid is simple cubic, and # actually corresponds to the # so-called 4x4x4 Monkhorst-Pack grid #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldfe 1.0d-6 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) ++=
, # repeated four times, with different shifts : shiftk # In cartesian coordinates, this grid is simple cubic, and # actually corresponds to the # so-called 4x4x4 Monkhorst-Pack grid #Definition of the SCF procedure nstep 10 # Maximal number of SCF cycles toldfe 1.0d-6 # Will stop when, twice in a row, the difference # between two consecutive evaluations of total energy # differ by less than toldfe (in Hartree) ++=")
31
iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor ETOT 1 -8.8611673348431 -8.861E+00 1.404E-03 6.305E+00 0.000E+00 0.000E+00 ETOT 2 -8.8661434670768 -4.976E-03 8.033E-07 1.677E-01 1.240E-30 1.240E-30 ETOT 3 -8.8662089742580 -6.551E-05 9.733E-07 4.402E-02 5.373E-30 4.959E-30 ETOT 4 -8.8662223695368 -1.340E-05 2.122E-08 4.605E-03 5.476E-30 5.166E-31 ETOT 5 -8.8662237078866 -1.338E-06 1.671E-08 4.634E-04 1.137E-30 6.199E-31 ETOT 6 -8.8662238907703 -1.829E-07 1.067E-09 1.326E-05 5.166E-31 5.166E-31 ETOT 7 -8.8662238959860 -5.216E-09 1.249E-10 3.283E-08 5.166E-31 0.000E+00 At SCF step 7, etot is converged : for the second time, diff in etot= 5.216E-09 < toldfe= 1.000E-06 cartesian forces (eV/Angstrom) at end: 1 0.00000000000000 0.00000000000000 0.00000000000000 2 0.00000000000000 0.00000000000000 0.00000000000000 Metals (T=0.25eV) ik=1 | eig [eV]: -5.8984 1.7993 1.9147 1.9147 2.8058 2.8058 141.3489 313.9870 313.9870 | focc: 2.0000 1.9999 1.9998 1.9998 1.9979 1.9979 0.0000 0.0000 0.0000
![iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor ETOT E E E E E+00 ETOT E E E E E-30 ETOT E E E E E-30 ETOT E E E E E-31 ETOT E E E E E-31 ETOT E E E E E-31 ETOT E E E E E+00 At SCF step 7, etot is converged : for the second time, diff in etot= 5.216E-09 < toldfe= 1.000E-06 cartesian forces (eV/Angstrom) at end: Metals (T=0.25eV) ik=1 | eig [eV]: | focc:](http://images.slideplayer.com/26/8287128/slides/slide_31.jpg "iter Etot(hartree) deltaE(h) residm vres2 diffor maxfor ETOT E E E E E+00 ETOT E E E E E-30 ETOT E E E E E-30 ETOT E E E E E-31 ETOT E E E E E-31 ETOT E E E E E-31 ETOT E E E E E+00 At SCF step 7, etot is converged : for the second time, diff in etot= 5.216E-09 < toldfe= 1.000E-06 cartesian forces (eV/Angstrom) at end: Metals (T=0.25eV) ik=1 | eig [eV]: | focc:")
37
DEPARTMENT OF PHYSICS AND DEPARTMENT OF NUCLEAR PHYSICS AND TECHNOLOGY, FACULTY OF ELECRICAL ENGINEERING AND INFORMATION TECHNOLOGY, SLOVAK UNIVERSITY OF TECHNOLOGY “Fe” RESULTS This work ab-initioExperiment f Ackland et al. potential EAM (nonmag. ) ab-initio (mag.) a BCC (Å)2.8662.831*2.88 c 2.872.8665 E COH (eV/atom)-4.2993-- c -4.28-4.316 Bulk Modulus (GPa) 179175.65*180 c 168.31.89 C`53.1457.73- c 59.40- C 44 83.56- a 142 d 112116 C 11 250.59252.62 a 250 d 242243.4 C 12 144.3137.16 a 145 d 145.6145 E VFA (eV)1.9112- b 1.93-2.02, *2.07 e 2.02±0.21.89 a FCC (Å)3.630---3.68 μ (μB)μ (μB)-2.19*2.31*2.22- E BCC – E FCC (eV)-0.0495---- * Fu CC, Williame F., Phys.Rev.Lett. 2004, 94, 175503 (a)Mehl MJ, Papaconstantopoulos DA, Yip S., editor. Handbook of materials modeling (b)Domain C., Becquart C., Phys.Rev. B 2002, 65, 024103 (c)Kittel C., Introduction to solid state physics, NY,Wiley, 1986 (d)Hirth JP, Lothe J., Theory of dislocation, 2.edition, NY, Wiley,1982 (e)Schepper LD et al., Phys.Rev. B, 1983, 27, 5257
 ab-initio (mag.) a BCC (Å) *2.88 c E COH (eV/atom) c Bulk Modulus (GPa) *180 c C` c C a 142 d C a 250 d C a 145 d E VFA (eV) b , *2.07 e 2.02± a FCC (Å) μ (μB)μ (μB)-2.19*2.31*2.22- E BCC – E FCC (eV) * Fu CC, Williame F., Phys.Rev.Lett. 2004, 94, (a)Mehl MJ, Papaconstantopoulos DA, Yip S., editor. Handbook of materials modeling (b)Domain C., Becquart C., Phys.Rev. B 2002, 65, (c)Kittel C., Introduction to solid state physics, NY,Wiley, 1986 (d)Hirth JP, Lothe J., Theory of dislocation, 2.edition, NY, Wiley,1982 (e)Schepper LD et al., Phys.Rev. B, 1983, 27,")
Similar presentations







 Workshop.>")





![DFT – Practice Simple Molecules & Solids [based on Chapters 5 & 2, Sholl & Steckel] Input files Supercells Molecules Solids.](/14/4353561/big_thumb.jpg "DFT – Practice Simple Molecules & Solids [based on Chapters 5 & 2, Sholl & Steckel] Input files Supercells Molecules Solids.>")
 www2.le.ac.uk/departments/physics/people/mervynroy.>")






! Hψ = Eψ A differential (operator) eigenvalue equation H.>")




