Download presentation
Presentation is loading. Please wait.

1
Ultra-large Multiple Sequence Alignment Tandy Warnow Founder Professor of Engineering The University of Illinois at Urbana-Champaign http://tandy.cs.illinois.edu

2
Orangutan GorillaChimpanzee Human From the Tree of the Life Website, University of Arizona Phylogeny (evolutionary tree)
")
3
Phylogenies and Applications Basic Biology: How did life evolve? Applications of phylogenies to: protein structure and function population genetics human migrations metagenomics

5
Computational Phylogenetics and Metagenomics Courtesy of the Tree of Life project

6
Hard Computational Problems NP-hard problems Large datasets 100,000+ sequences thousands of genes “Big data” complexity: model misspecification fragmentary sequences errors in input data streaming data

7
Warnow Research Goal: improve accuracy, speed, robustness, or mathematical guarantees of computational methods, to enable highly accurate analyses of real datasets Techniques: divide-and-conquer, iteration, chordal graph theory, and probability theory Evaluation: synthetic and real data; collaborations with biologists and linguists Examples: Historical linguistics, 1994-present Absolute fast converging methods 1997-2002 Phylogenetic networks, 2003-2005 Genome rearrangements, 2000-2006 Multiple sequence alignment, 2009-present (many papers, including SATé-1 (Science), SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)
, SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)")
8
Warnow Research Goal: improve accuracy, speed, robustness, or mathematical guarantees of computational methods, to enable highly accurate analyses of real datasets Techniques: divide-and-conquer, iteration, chordal graph theory, and probability theory Evaluation: synthetic and real data; collaborations with biologists and linguists Examples: Historical linguistics, 1994-present Absolute fast converging methods 1997-2002 Phylogenetic networks, 2003-2005 Genome rearrangements, 2000-2006 Multiple sequence alignment, 2009-present (many papers, including SATé-1 (Science), SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)
, SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)")
9
Warnow Research Goal: improve accuracy, speed, robustness, or mathematical guarantees of computational methods, to enable highly accurate analyses of real datasets Techniques: divide-and-conquer, iteration, chordal graph theory, and probability theory Evaluation: synthetic and real data; collaborations with biologists and linguists Examples: Historical linguistics, 1994-present Absolute fast converging methods 1997-2002 Phylogenetic networks, 2003-2005 Genome rearrangements, 2000-2006 Multiple sequence alignment, 2009-present (many papers, including SATé-1 (Science), SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)
, SATé-2 (Syst Biol), PASTA (RECOMB and J Comp Biol), and UPP (Genome Biology)) Supertree methods, 2009-present Metagenomic analysis, 2014-present Coalescent-based species tree estimation (2011-present, including Science 2014a, Science 2014b, PNAS 2014)")
10
DNA Sequence Evolution AAGACTT TGGACTTAAGGCCT -3 mil yrs -2 mil yrs -1 mil yrs today AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT TAGCCCATAGACTTAGCGCTTAGCACAAAGGGCAT TAGCCCTAGCACTT AAGACTT TGGACTTAAGGCCT AGGGCATTAGCCCTAGCACTT AAGGCCTTGGACTT AGCGCTTAGCACAATAGACTTTAGCCCAAGGGCAT

11
Phylogeny Problem TAGCCCATAGACTTTGCACAATGCGCTTAGGGCAT UVWXY U VW X Y

12
Markov Model of Site Evolution Simplest (Jukes-Cantor, 1969): The model tree T is binary and has substitution probabilities p(e) on each edge e. The state at the root is randomly drawn from {A,C,T,G} (nucleotides) If a site (position) changes on an edge, it changes with equal probability to each of the remaining states. The evolutionary process is Markovian. More complex models (such as the General Time Reversible model, or the General Markov model) are also considered, often with little change to the theory.
 If a site (position) changes on an edge, it changes with equal probability to each of the remaining states. The evolutionary process is Markovian. More complex models (such as the General Time Reversible model, or the General Markov model) are also considered, often with little change to the theory..")
13
Quantifying Error FN: false negative (missing edge) FP: false positive (incorrect edge) FN FP 50% error rate
 FP: false positive (incorrect edge) FN FP 50% error rate")
14
Statistical Consistency error Data Maximum likelihood is statistically consistent under standard models (e.g., GTR)
")
15
Mathematical Questions Is the model tree identifiable? Which estimation methods are statistically consistent under this model? How much data does the method need to estimate the model tree correctly (with high probability)? What is the impact of model misspecification? What is the computational complexity of an estimation problem?
. What is the impact of model misspecification. What is the computational complexity of an estimation problem .")
16
The Classical Phylogeny Problem TAGCCCATAGACTTTGCACAATGCGCTTAGGGCAT UVWXY U VW X Y

17
TAGCCCATAGACTTTGCACAATGCGCTTAGGGCAT UVWXY U VW X Y Much is known about this problem from a mathematical and empirical viewpoint

18
AGAT TAGACTTTGCACAATGCGCTT AGGGCATGA UVWXY U VW X Y However…

19
…ACGGTGCAGTTACCA… MutationDeletion …ACCAGTCACCA… Indels (insertions and deletions)
")
20
…ACGGTGCAGTTACC-A… …AC----CAGTCACCTA… The true multiple alignment – Reflects historical substitution, insertion, and deletion events – Defined using transitive closure of pairwise alignments computed on edges of the true tree … ACGGTGCAGTTACCA … Substitution Deletion … ACCAGTCACCTA … Insertion

21
Input: unaligned sequences S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA

22
Phase 1: Alignment S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA

23
Phase 2: Construct tree S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA S1 S4 S2 S3

24
Phylogenomic pipeline Select taxon set and markers Gather and screen sequence data, possibly identify orthologs Compute multiple sequence alignments for each locus, and construct gene trees Compute species tree or network: – Combine the estimated gene trees, OR – Estimate a tree from a concatenation of the multiple sequence alignments Get statistical support on each branch (e.g., bootstrapping) Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology
 Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology")
25
Phylogenomic pipeline Select taxon set and markers Gather and screen sequence data, possibly identify orthologs Compute multiple sequence alignments for each locus, and construct gene trees Compute species tree or network: – Combine the estimated gene trees, OR – Estimate a tree from a concatenation of the multiple sequence alignments Get statistical support on each branch (e.g., bootstrapping) Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology Coalescent-based species tree estimation!
 Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology Coalescent-based species tree estimation!")
26
Phylogenomic pipeline Select taxon set and markers Gather and screen sequence data, possibly identify orthologs Compute multiple sequence alignments for each locus, and construct gene trees Compute species tree or network: – Combine the estimated gene trees, OR – Estimate a tree from a concatenation of the multiple sequence alignments Get statistical support on each branch (e.g., bootstrapping) Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology
 Estimate dates on the nodes of the phylogeny Use species tree with branch support and dates to understand biology")
27
Large-scale Alignment Estimation Many genes are considered unalignable due to high rates of evolution Only a few methods can analyze large datasets iPlant (NSF Plant Biology Collaborative) and other projects planning to construct phylogenies with 500,000 taxa
 and other projects planning to construct phylogenies with 500,000 taxa")
28
1kp: Thousand Transcriptome Project First study (Wickett, Mirarab, et al., PNAS 2014) had ~100 species and ~800 genes, gene trees and alignments estimated using SATé, and a coalescent-based species tree estimated using ASTRAL Second study: Plant Tree of Life based on transcriptomes of ~1200 species, and more than 13,000 gene families (most not single copy) Gene Tree Incongruence G. Ka-Shu Wong U Alberta N. Wickett Northwestern J. Leebens-Mack U Georgia N. Matasci iPlant T. Warnow, S. Mirarab, N. Nguyen, UIUC UT-Austin UT-Austin Challenges: Species tree estimation from conflicting gene trees Gene tree estimation of datasets with > 100,000 sequences Plus many many other people…

29
Multiple Sequence Alignment (MSA): a scientific grand challenge 1 S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- … Sn = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC … Sn = TCACGACCGACA Novel techniques needed for scalability and accuracy NP-hard problems and large datasets Current methods do not provide good accuracy Few methods can analyze even moderately large datasets Many important applications besides phylogenetic estimation 1 Frontiers in Massive Data Analysis, National Academies Press, 2013
: a scientific grand challenge 1 S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT GACCGC-- … Sn = TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC … Sn = TCACGACCGACA Novel techniques needed for scalability and accuracy NP-hard problems and large datasets Current methods do not provide good accuracy Few methods can analyze even moderately large datasets Many important applications besides phylogenetic estimation 1 Frontiers in Massive Data Analysis, National Academies Press, 2013")
30
This talk “Big data” multiple sequence alignment SATé (Science 2009, Systematic Biology 2012) and PASTA (RECOMB and J Comp Biol 2015), methods for co-estimation of alignments and trees UPP (Genome Biology 2015): ultra-large multiple sequence alignment, using the “Ensemble of HMMs technique”. Other applications of the eHMM technique: –phylogenetic placement (SEPP, PSB 2012) –metagenomic taxon identification (TIPP, Bioinformatics 2014) –protein structure and function classification –gene binning
 –metagenomic taxon identification (TIPP, Bioinformatics 2014) –protein structure and function classification –gene binning.")
31
Multiple Sequence Alignment

32
First Align, then Compute the Tree S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA S1 S4 S2 S3

33
S1 = -AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA S1 S4 S2 S3 Co-estimation would be much better!!!

34
Simulation Studies S1S2 S3S4 S1 = - AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-CT-------GACCGC-- S4 = -------TCAC--GACCGACA S1 = AGGCTATCACCTGACCTCCA S2 = TAGCTATCACGACCGC S3 = TAGCTGACCGC S4 = TCACGACCGACA S1 = - AGGCTATCACCTGACCTCCA S2 = TAG-CTATCAC--GACCGC-- S3 = TAG-C--T-----GACCGC-- S4 = T---C-A-CGACCGA----CA Compare True tree and alignment S1S4 S3S2 Estimated tree and alignment Unaligned Sequences

35
Quantifying Error FN: false negative (missing edge) FP: false positive (incorrect edge) 50% error rate FN FP
 FP: false positive (incorrect edge) 50% error rate FN FP")
36
Two-phase estimation Alignment methods Clustal POY (and POY*) Probcons (and Probtree) Probalign MAFFT Muscle Di-align T-Coffee Prank (PNAS 2005, Science 2008) Opal (ISMB and Bioinf. 2007) FSA (PLoS Comp. Bio. 2009) Infernal (Bioinf. 2009) Etc. Phylogeny methods Bayesian MCMC Maximum parsimony Maximum likelihood Neighbor joining FastME UPGMA Quartet puzzling Etc. RAxML: heuristic for large-scale ML optimization
 FSA (PLoS Comp. Bio. 2009) Infernal (Bioinf. 2009) Etc. Phylogeny methods Bayesian MCMC Maximum parsimony Maximum likelihood Neighbor joining FastME UPGMA Quartet puzzling Etc. RAxML: heuristic for large-scale ML optimization.")
37
1000-taxon models, ordered by difficulty (Liu et al., 2009)
")
38
SATé “Family” of methods Iterative divide-and-conquer methods – Each iteration re-aligns the sequences using the current tree, running preferred MSA methods on small local subsets, and merging subset alignments – Each iteration computes an ML tree on the current alignment, under the GTR (Generalized Time Reversible) Markov model of evolution Note: these methods are “MSA boosters”, designed to improve accuracy and/or scalability of the base method We show results using MAFFT-l-ins-i to align subsets
 Markov model of evolution Note: these methods are MSA boosters , designed to improve accuracy and/or scalability of the base method We show results using MAFFT-l-ins-i to align subsets")
39
Re-aligning on a tree A B D C Merge sub- alignments Estimate ML tree on merged alignment Decompose dataset AB CD Align subsets AB CD ABCD

40
SATé and PASTA Algorithms Estimate ML tree on new alignment Tree Obtain initial alignment and estimated ML tree Use tree to compute new alignment Alignment Repeat until termination condition, and return the alignment/tree pair with the best ML score

41
1000-taxon models, ordered by difficulty – rate of evolution generally increases from left to right SATé-1 24 hour analysis, on desktop machines (Similar improvements for biological datasets) SATé-1 can analyze up to about 8,000 sequences. SATé-1 (Science 2009) performance
 performance.")
42
1000-taxon models ranked by difficulty SATé-1 and SATé-2 (Systematic Biology, 2012) SATé-1: up to 8K SATé-2: up to ~50K
 SATé-1: up to 8K SATé-2: up to ~50K")
43
SATé variants differ only in the decomposition strategy A B D C Merge sub- alignments Estimate ML tree on merged alignment Decompose dataset AB CD Align subsets AB CD ABCD

44
SATé-II: centroid edge decomposition ABCDE ABC AB AB C DE DE SATé-II makes all subsets small (user parameter), and can analyze 50K sequences, SATé-I decomposition produced clades and had bigger subsets; limited to 8K sequences
, and can analyze 50K sequences, SATé-I decomposition produced clades and had bigger subsets; limited to 8K sequences")
45
SATé: merger strategy ABCDE ABC AB AB C DE DE Both SATé’s use the same hierarchical merger strategy. On large (50K) datasets, the last pairwise merger can use more than 70% of the running time
 datasets, the last pairwise merger can use more than 70% of the running time.")
46
PASTA merging: Step 1 Compute a spanning tree connecting alignment subsets

47
PASTA merging: Step 2 AB BD CD DE AB BD CD DE Use Opal (or Muscle) to merge adjacent subset alignments in the spanning tree
 to merge adjacent subset alignments in the spanning tree")
48
PASTA merging: Step 3 Use transitivity to merge all pairwise-merged alignments from Step 2 into final an alignment on entire dataset AB + BD = ABD ABD + CD = ABCD ABCD + DE = ABCDE AB BD CD DE Overall: O(n log(n) + L)
 + L)")
49
PASTA vs. SATé-II profiling and scaling

50
PASTA Running Time and Scalability One iteration Using 12 cpus 1 node on Lonestar TACC Maximum 24 GB memory Showing wall clock running time ~ 1 hour for 10k taxa ~ 17 hours for 200k taxa

52
PASTA vs. SATé-II Difference is how subset alignments are merged together (transitivity instead of Opal/Muscle). As expected, PASTA is faster and can analyze larger datasets. Unexpected: PASTA produces more accurate alignments and trees (on both simulated and biological data, including DNA, RNA, and AA sequences). Thus, transitivity applied to compatible and overlapping alignments gives a surprisingly accurate technique for merging a collection of alignments.
. As expected, PASTA is faster and can analyze larger datasets. Unexpected: PASTA produces more accurate alignments and trees (on both simulated and biological data, including DNA, RNA, and AA sequences). Thus, transitivity applied to compatible and overlapping alignments gives a surprisingly accurate technique for merging a collection of alignments..")
53
PASTA and SATé-II: MSA “boosters” PASTA and SATé-II are techniques for improving the scalability of MSA methods to large datasets. We showed results here using MAFFT-l-ins-i to align small subsets with 200 sequences. We have also explored results using other MSA methods (e.g., Prank, Clustal, Bali-Phy), and obtain similar improvements in accuracy and/or scalability.
, and obtain similar improvements in accuracy and/or scalability..")
54
1kp: Thousand Transcriptome Project Plant Tree of Life based on transcriptomes of ~1200 species More than 13,000 gene families (most not single copy) Gene Tree Incongruence G. Ka-Shu Wong U Alberta N. Wickett Northwestern J. Leebens-Mack U Georgia N. Matasci iPlant T. Warnow, S. Mirarab, N. Nguyen, UIUC UT-Austin UT-Austin Challenge: Massive gene tree conflict consistent with ILS Alignment of datasets with > 100,000 sequences Plus many many other people…

55
1KP dataset: more than 100,000 p450 amino-acid sequences, many fragmentary

56
1KP dataset: more than 100,000 p450 amino-acid sequences, many fragmentary All standard multiple sequence alignment methods we tested performed poorly on datasets with fragments.

57
1kp: Thousand Transcriptome Project Plant Tree of Life based on transcriptomes of ~1200 species More than 13,000 gene families (most not single copy) Gene Tree Incongruence G. Ka-Shu Wong U Alberta N. Wickett Northwestern J. Leebens-Mack U Georgia N. Matasci iPlant T. Warnow, S. Mirarab, N. Nguyen UIUCUT-Austin UT-Austin Challenge: Alignment of datasets with > 100,000 sequences with many fragmentary sequences Plus many many other people…

58
UPP UPP = “Ultra-large multiple sequence alignment using Phylogeny-aware Profiles” Nguyen, Mirarab, and Warnow. Genome Biology, 2014. Purpose: highly accurate large-scale multiple sequence alignments, even in the presence of fragmentary sequences.

59
UPP UPP = “Ultra-large multiple sequence alignment using Phylogeny-aware Profiles” Nguyen, Mirarab, and Warnow. Genome Biology, 2014. Purpose: highly accurate large-scale multiple sequence alignments, even in the presence of fragmentary sequences. Uses an ensemble of HMMs

60
Simple idea (not UPP) Select random subset of sequences, and build “backbone alignment” Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM
 Select random subset of sequences, and build backbone alignment Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM")
61
One Hidden Markov Model for the entire alignment?

62
Simple idea (not UPP) Select random subset of sequences, and build “backbone alignment” Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM
 Select random subset of sequences, and build backbone alignment Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM")
63
Select random subset of sequences, and build “backbone alignment” Construct a Hidden Markov Model (HMM) on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM This approach works well if the dataset is small and has low evolutionary rates, but is not very accurate otherwise.
 on the backbone alignment Add all remaining sequences to the backbone alignment using the HMM This approach works well if the dataset is small and has low evolutionary rates, but is not very accurate otherwise.")
64
One Hidden Markov Model for the entire alignment? HMM 1

65
Or 2 HMMs? HMM 1 HMM 2

66
HMM 1 HMM 3 HMM 4 HMM 2 Or 4 HMMs?

67
m HMM 2 HMM 3 HMM 1 HMM 4 HMM 5 HMM 6 HMM 7 Or all 7 HMMs?

68
UPP Algorithmic Approach 1.Select random subset of full-length sequences, and build “backbone alignment” 2.Construct an “Ensemble of Hidden Markov Models” on the backbone alignment 3.Add all remaining sequences to the backbone alignment using the Ensemble of HMMs

69
UPP Algorithmic Approach 1.Select random subset of full-length sequences, and build “backbone alignment” and “backbone tree” Notes: – Need to avoid fragments in the backbone – We show results using PASTA for the backbone alignment and tree, but are other methods can be used – we have explored BAli-Phy, a powerful Bayesian statistical method – Random is good when taxonomic sampling is relatively uniform, but directed sampling can improve accuracy – We explored backbones with 100 and 1000 sequences, even when the full dataset is very big (1,000,000 – one million)
")
70
UPP Algorithmic Approach 2.Construct an “Ensemble of Hidden Markov Models” on the backbone alignment – Technique: Create set of subsets (using the tree). Then, for each subset, build an HMM on the induced alignment on each subset. – Note: Different subset sizes are good for different situations, and the ensemble technique is more accurate than disjoint sets

71
UPP Algorithmic Approach 3.Add all remaining sequences to the backbone alignment using the Ensemble of HMMs – For each of the remaining sequences s, find H, the HMM from the ensemble that has the best score (i.e., HMM maximizing Pr(s|H)) – Use HMMER code and H to add s into the backbone alignment
) – Use HMMER code and H to add s into the backbone alignment")
72
Evaluation Simulated datasets (some have fragmentary sequences): – 10K to 1,000,000 sequences in RNASim – complex RNA sequence evolution simulation – 1000-sequence nucleotide datasets from SATé papers – 5000-sequence AA datasets (from FastTree paper) – 10,000-sequence Indelible nucleotide simulation Biological datasets: – Proteins: largest BaliBASE and HomFam – RNA: 3 CRW datasets up to 28,000 sequences
: – 10K to 1,000,000 sequences in RNASim – complex RNA sequence evolution simulation – 1000-sequence nucleotide datasets from SATé papers – 5000-sequence AA datasets (from FastTree paper) – 10,000-sequence Indelible nucleotide simulation Biological datasets: – Proteins: largest BaliBASE and HomFam – RNA: 3 CRW datasets up to 28,000 sequences")
73
RNASim: alignment error Note: Mafft was run under default settings for 10K and 50K sequences and under Parttree for 100K sequences, and fails to complete under any setting For 200K sequences. Clustal-Omega only completes on 10K dataset. All methods given 24 hrs on a 12-core machine

74
RNASim: tree error Note: Mafft was run under default settings for 10K and 50K sequences and under Parttree for 100K sequences, and fails to complete under any setting For 200K sequences. Clustal-Omega only completes on 10K dataset. All methods given 24 hrs on a 12-core machine

75
RNASim Million Sequences: alignment error Notes: We show alignment error using average of SP-FN and SP-FP. UPP variants have better alignment scores than PASTA. (Not shown: Total Column Scores – PASTA more accurate than UPP) No other methods tested could complete on these data PASTA under-aligns: its alignment is 43 times wider than true alignment (~900 Gb of disk space). UPP alignments were closer in length to true alignment (0.93 to 1.38 wider).
 No other methods tested could complete on these data PASTA under-aligns: its alignment is 43 times wider than true alignment (~900 Gb of disk space). UPP alignments were closer in length to true alignment (0.93 to 1.38 wider)..")
76
RNASim Million Sequences: tree error Using 12 processors: UPP(Fast,NoDecomp) took 2.2 days, UPP(Fast) took 11.9 days, and PASTA took 10.3 days
 took 2.2 days, UPP(Fast) took 11.9 days, and PASTA took 10.3 days")
77
Performance on fragmentary datasets of the 1000M2 model condition UPP vs. PASTA: impact of fragmentation Under high rates of evolution, PASTA is badly impacted by fragmentary sequences (the same is true for other methods). Under low rates of evolution, PASTA can still be highly accurate (data not shown). UPP continues to have good accuracy even on datasets with many fragments under all rates of evolution.
. Under low rates of evolution, PASTA can still be highly accurate (data not shown). UPP continues to have good accuracy even on datasets with many fragments under all rates of evolution..")
78
UPP Running Time Wall-clock time used (in hours) given 12 processors
 given 12 processors")
79
Current Related Research UPP: Using iteration Using other MSA models (not just HMMs) within the “Ensemble” Using structural alignments for the backbone Using powerful statistical methods to produce the backbone alignment and tree PASTA : Using powerful statistical methods for the subset alignments Improving the pairwise merging technique ENSEMBLE OF HMMS: Metagenomic taxon identification (collaboration with Mihai Pop, Maryland) Gene binning (joint with Jian Peng, UIUC, and with Jim Leebens-Mack, Georgia) Protein structure and function classification (collaborations with Martin Weigt, Paris, and Jian Peng, UIUC)
 within the Ensemble Using structural alignments for the backbone Using powerful statistical methods to produce the backbone alignment and tree PASTA : Using powerful statistical methods for the subset alignments Improving the pairwise merging technique ENSEMBLE OF HMMS: Metagenomic taxon identification (collaboration with Mihai Pop, Maryland) Gene binning (joint with Jian Peng, UIUC, and with Jim Leebens-Mack, Georgia) Protein structure and function classification (collaborations with Martin Weigt, Paris, and Jian Peng, UIUC)")
80
Acknowledgments PhD students: Nam Nguyen (now postdoc at UIUC) and Siavash Mirarab (now faculty at UCSD) Undergrad: Keerthana Kumar Current NSF grants: ABI-1458652 (multiple sequence alignment) III:AF:1513629 (metagenomics – collaborative with Mihai Pop, University of Maryland) DBI:1461364 (phylogenomics – collaborative with Rice and Stanford) CCF:1535977 (graph algorithms to improve phylogenetic estimation – collaborative with Berkeley) Other recent or current support: Guggenheim Foundation, NSF DEB:0733029, NSF DBI:1062335, Microsoft Research New England, David Bruton Jr. Centennial Professorship, TACC (Texas Advanced Computing Center), the University of Alberta (Canada), Grainger Foundation (at UIUC), and UIUC TACC, UTCS, and UIUC computational resources
, the University of Alberta (Canada), Grainger Foundation (at UIUC), and UIUC TACC, UTCS, and UIUC computational resources.")
81
Alignment Accuracy – Correct columns

82
TIPP: high accuracy taxonomic identification of metagenomic data TIPP: taxon identification and phylogenetic profiling (Bioinformatics, 2014) Technique: combines UPP alignments and phylogenetic placement algorithms, and considers statistical uncertainty. Results: better accuracy than all current methods, even for sequencing technologies producing high indel rates Research funded by new NSF grant III:AF:1513629 (collaborative with Mihai Pop at University of Maryland)
.")
83
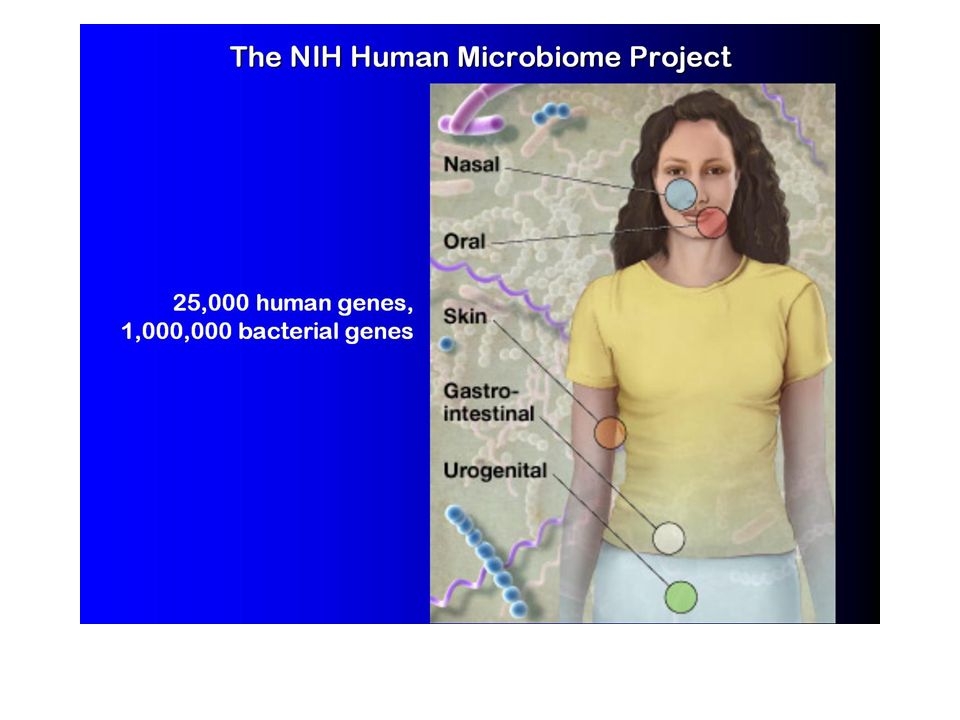
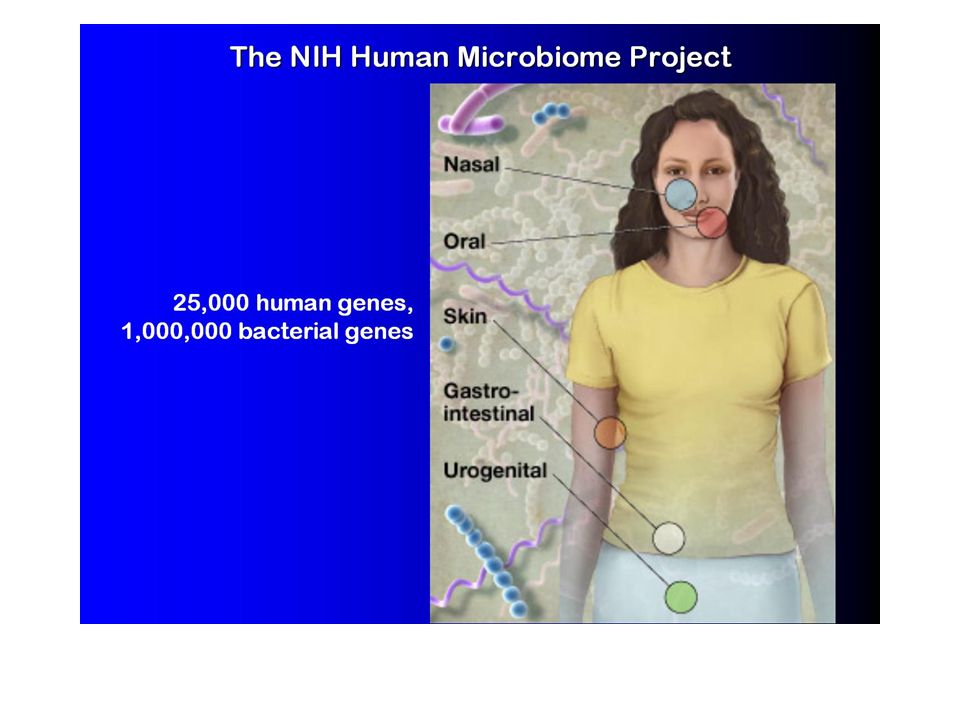
Metagenomic taxonomic identification and phylogenetic profiling Metagenomics, Venter et al., Exploring the Sargasso Sea: Scientists Discover One Million New Genes in Ocean Microbes

84
1. What is this fragment? (Classify each fragment as well as possible.) 2. What is the taxonomic distribution in the dataset? (Note: helpful to use marker genes.) 3. What are the organisms in this metagenomic sample doing together? Basic Questions
 3. What are the organisms in this metagenomic sample doing together. Basic Questions.")
85
Scientific challenges: Ultra-large multiple-sequence alignment Alignment-free phylogeny estimation Supertree estimation Estimating species trees from many gene trees Genome rearrangement phylogeny Reticulate evolution Visualization of large trees and alignments Data mining techniques to explore multiple optima Theoretical guarantees under Markov models of evolution Techniques: machine learning, applied probability theory, graph theory, combinatorial optimization, supercomputing, and heuristics The Tree of Life: Multiple Challenges

86
High indel datasets containing known genomes Note: NBC, MetaPhlAn, and MetaPhyler cannot classify any sequences from at least one of the high indel long sequence datasets, and mOTU terminates with an error message on all the high indel datasets.

87
TIPP vs. other abundance profilers TIPP is highly accurate, even in the presence of high indel rates and novel genomes, and for both short and long reads. All other methods have some vulnerability (e.g., mOTU is only accurate for short reads and is impacted by high indel rates).
..")
88
Metagenomic Taxon Identification Objective: classify short reads in a metagenomic sample

89
Objective: Distribution of the species (or genera, or families, etc.) within the sample. For example: The distribution of the sample at the species-level is: 50% species A 20% species B 15% species C 14% species D 1% species E Abundance Profiling

90
Objective: Distribution of the species (or genera, or families, etc.) within the sample. Leading techniques: PhymmBL (Brady & Salzberg, Nature Methods 2009) NBC (Rosen, Reichenberger, and Rosenfeld, Bioinformatics 2011) MetaPhyler (Liu et al., BMC Genomics 2011), from the Pop lab at the University of Maryland MetaPhlAn (Segata et al., Nature Methods 2012), from the Huttenhower Lab at Harvard mOTU (Bork et al., Nature Methods 2013) MetaPhyler, MetaPhlAn, and mOTU are marker-based techniques (but use different marker genes). Marker gene are single-copy, universal, and resistant to horizontal transmission. Abundance Profiling
 NBC (Rosen, Reichenberger, and Rosenfeld, Bioinformatics 2011) MetaPhyler (Liu et al., BMC Genomics 2011), from the Pop lab at the University of Maryland MetaPhlAn (Segata et al., Nature Methods 2012), from the Huttenhower Lab at Harvard mOTU (Bork et al., Nature Methods 2013) MetaPhyler, MetaPhlAn, and mOTU are marker-based techniques (but use different marker genes). Marker gene are single-copy, universal, and resistant to horizontal transmission. Abundance Profiling.")
91
“Novel” genome datasets Note: mOTU terminates with an error message on the long fragment datasets and high indel datasets.

92
Summary SATé-1 (Science 2009), SATé-2 (Systematic Biology 2012), co-estimation of alignments and trees. SATé-2 is well established in the biology community. PASTA (RECOMB 2014 and J Comp Biol 2015) is the replacement for SATé. PASTA can analyze up to 1,000,000 sequences. UPP (ultra-large multiple sequence alignment), Genome Biology 2015. Uses a collection of HMMs to represent a “backbone alignment”. Improves alignment and also detection of remote homology compared to a single HMM. UPP produces highly accurate alignments, even in the presence of fragmentary sequences. Can analyze datasets with 1,000,000 sequences. Other applications of the Ensemble of HMMs technique TIPP (metagenomic taxon identification and abundance profiling), Bioinformatics 2014. SEPP (phylogenetic placement), PSB 2012. Protein sequence analysis (collaborations with Martin Weigt, Paris, and Jian Peng, UIUC)
 is the replacement for SATé. PASTA can analyze up to 1,000,000 sequences. UPP (ultra-large multiple sequence alignment), Genome Biology Uses a collection of HMMs to represent a backbone alignment . Improves alignment and also detection of remote homology compared to a single HMM. UPP produces highly accurate alignments, even in the presence of fragmentary sequences. Can analyze datasets with 1,000,000 sequences. Other applications of the Ensemble of HMMs technique TIPP (metagenomic taxon identification and abundance profiling), Bioinformatics SEPP (phylogenetic placement), PSB Protein sequence analysis (collaborations with Martin Weigt, Paris, and Jian Peng, UIUC).")
93
SEPP SEPP: SATé-enabled Phylogenetic Placement, by Mirarab, Nguyen, and Warnow. Pacific Symposium on Biocomputing, 2012, special session on the Human Microbiome Objective: –phylogenetic analysis of single-gene datasets with fragmentary sequences Introduces “HMM Family” technique

94
Phylogenetic Placement ACT..TAGA..AAGC...ACATAGA...CTTTAGC...CCAAGG...GCAT ACCG CGAG CGG GGCT TAGA GGGGG TCGAG GGCG GGG. ACCT Fragmentary sequences from some gene Full-length sequences for same gene, and an alignment and a tree

95
Step 1: Align each query sequence to backbone alignment Step 2: Place each query sequence into backbone tree, using extended alignment Phylogenetic Placement

96
HMMER vs. PaPaRa Alignments Increasing rate of evolution 0.0

97
Align Sequence S1 S4 S2 S3 S1 = -AGGCTATCACCTGACCTCCA-AA S2 = TAG-CTATCAC--GACCGC--GCA S3 = TAG-CT-------GACCGC--GCT S4 = TAC----TCAC--GACCGACAGCT Q1 = TAAAAC

98
Align Sequence S1 S4 S2 S3 S1 = -AGGCTATCACCTGACCTCCA-AA S2 = TAG-CTATCAC--GACCGC--GCA S3 = TAG-CT-------GACCGC--GCT S4 = TAC----TCAC--GACCGACAGCT Q1 = -------T-A--AAAC--------

99
Phylogenetic Placement Align each query sequence to backbone alignment –HMMALIGN (Eddy, Bioinformatics 1998) –PaPaRa (Berger and Stamatakis, Bioinformatics 2011) Place each query sequence into backbone tree –Pplacer (Matsen et al., BMC Bioinformatics, 2011) –EPA (Berger and Stamatakis, Systematic Biology 2011) Note: pplacer and EPA use maximum likelihood, and are reported to have the same accuracy.
 –PaPaRa (Berger and Stamatakis, Bioinformatics 2011) Place each query sequence into backbone tree –Pplacer (Matsen et al., BMC Bioinformatics, 2011) –EPA (Berger and Stamatakis, Systematic Biology 2011) Note: pplacer and EPA use maximum likelihood, and are reported to have the same accuracy.")
100
Place Sequence S1 S4 S2 S3 Q1 S1 = -AGGCTATCACCTGACCTCCA-AA S2 = TAG-CTATCAC--GACCGC--GCA S3 = TAG-CT-------GACCGC--GCT S4 = TAC----TCAC--GACCGACAGCT Q1 = -------T-A--AAAC--------

101
HMMER+pplacer: 1) build one HMM for the entire alignment 2) Align fragment to the HMM, and insert into alignment 3) Insert fragment into tree to optimize likelihood
 build one HMM for the entire alignment 2) Align fragment to the HMM, and insert into alignment 3) Insert fragment into tree to optimize likelihood")
102
Using SEPP for taxon identification ACT..TAGA..AAGC...ACATAGA...CTTTAGC...CCAAGG...GCAT ACCG CGAG CGG GGCT TAGA GGGGG TCGAG GGCG GGG. ACCT Fragmentary sequences from some gene Full-length sequences for same gene, and an alignment and a tree

103
Using SEPP for taxon identification ACT..TAGA..AAGC...ACATAGA...CTTTAGC...CCAAGG...GCAT ACCG CGAG CGG GGCT TAGA GGGGG TCGAG GGCG GGG. ACCT Fragmentary sequences from some gene Full-length sequences for same gene, and an alignment and a tree

104
SEPP(10%), based on ~10 HMMs 0.0 Increasing rate of evolution
, based on ~10 HMMs 0.0 Increasing rate of evolution")
106
SEPP produced more accurate phylogenetic placements than HMMER+pplacer. The only difference is the use of a Family of HMMs instead of one HMM. The biggest differences are for datasets with high rates of evolution. SEPP vs. HMMER+pplacer

107
Scientific challenges: Ultra-large multiple-sequence alignment Gene tree estimation Metagenomic classification Alignment-free phylogeny estimation Supertree estimation Estimating species trees from many gene trees Genome rearrangement phylogeny Reticulate evolution Visualization of large trees and alignments Data mining techniques to explore multiple optima Theoretical guarantees under Markov models of evolution Techniques: applied probability theory, graph theory, supercomputing, and heuristics Testing: simulations and real data The Tree of Life: Multiple Challenges

108
Scientific challenges: Ultra-large multiple-sequence alignment Gene tree estimation Metagenomic classification Alignment-free phylogeny estimation Supertree estimation Estimating species trees from many gene trees Genome rearrangement phylogeny Reticulate evolution Visualization of large trees and alignments Data mining techniques to explore multiple optima Theoretical guarantees under Markov models of evolution Techniques: applied probability theory, graph theory, supercomputing, and heuristics Testing: simulations and real data The Tree of Life: Multiple Challenges

109
SATé-II running time profiling

110
Figure from Mirarab et al., J. Computational Biology 2014

Similar presentations