Download presentation
Presentation is loading. Please wait.

1
Combinatorially Complex Equilibrium Model Selection Tom Radivoyevitch Assistant Professor Epidemiology and Biostatistics Case Western Reserve University Email: txr24@case.edutxr24@case.edu Website: http://epbi-radivot.cwru.edu/http://epbi-radivot.cwru.edu/

2
Combinatorially Complex Equilibrium Model Selection (overview/title breakdown) Combinatorially Complex Equilibria: Combinatorially Complex Equilibria: few reactants => many possible complexes Generate the possible models + Select or average the best Generate the possible models + Select or average the best Selection/Averaging: Akaike Information Criterion (AIC) Selection/Averaging: Akaike Information Criterion (AIC) AIC decreases with P and then increases AIC decreases with P and then increases Billions of models, but only thousands near AIC upturn Billions of models, but only thousands near AIC upturn Challenge: Generate 1P, 2P, 3P model space chunks sequentially Challenge: Generate 1P, 2P, 3P model space chunks sequentially 8GB RAM => max of ~5000 models at a time 8GB RAM => max of ~5000 models at a time Models = Hypotheses Models = Hypotheses Spurs: complete K = ∞ [Complex]=0 Spurs: complete K = ∞ [Complex]=0 Grids: binary K equal each other Grids: binary K equal each other R package ccems (CRAN) implements the methods R package ccems (CRAN) implements the methods
![Combinatorially Complex Equilibrium Model Selection (overview/title breakdown) Combinatorially Complex Equilibria: Combinatorially Complex Equilibria: few reactants => many possible complexes Generate the possible models + Select or average the best Generate the possible models + Select or average the best Selection/Averaging: Akaike Information Criterion (AIC) Selection/Averaging: Akaike Information Criterion (AIC) AIC decreases with P and then increases AIC decreases with P and then increases Billions of models, but only thousands near AIC upturn Billions of models, but only thousands near AIC upturn Challenge: Generate 1P, 2P, 3P model space chunks sequentially Challenge: Generate 1P, 2P, 3P model space chunks sequentially 8GB RAM => max of ~5000 models at a time 8GB RAM => max of ~5000 models at a time Models = Hypotheses Models = Hypotheses Spurs: complete K = ∞ [Complex]=0 Spurs: complete K = ∞ [Complex]=0 Grids: binary K equal each other Grids: binary K equal each other R package ccems (CRAN) implements the methods R package ccems (CRAN) implements the methods](http://images.slideplayer.com/25/8038000/slides/slide_2.jpg "Combinatorially Complex Equilibrium Model Selection (overview/title breakdown) Combinatorially Complex Equilibria: Combinatorially Complex Equilibria: few reactants => many possible complexes Generate the possible models + Select or average the best Generate the possible models + Select or average the best Selection/Averaging: Akaike Information Criterion (AIC) Selection/Averaging: Akaike Information Criterion (AIC) AIC decreases with P and then increases AIC decreases with P and then increases Billions of models, but only thousands near AIC upturn Billions of models, but only thousands near AIC upturn Challenge: Generate 1P, 2P, 3P model space chunks sequentially Challenge: Generate 1P, 2P, 3P model space chunks sequentially 8GB RAM => max of ~5000 models at a time 8GB RAM => max of ~5000 models at a time Models = Hypotheses Models = Hypotheses Spurs: complete K = ∞ [Complex]=0 Spurs: complete K = ∞ [Complex]=0 Grids: binary K equal each other Grids: binary K equal each other R package ccems (CRAN) implements the methods R package ccems (CRAN) implements the methods")
3
Center Relevance Structures constrain minimal simplicity and maximal complexity of possible complexes and models in the model space Structures constrain minimal simplicity and maximal complexity of possible complexes and models in the model space Move prior knowledge of structures into enzyme models Move prior knowledge of structures into enzyme models System Biology models integrate enzyme models System Biology models integrate enzyme models Protein-Protein Protein-Ligand Interaction readouts: Protein-Protein Protein-Ligand Interaction readouts: Enzyme activities and DLS mass (i.e. average measures) Enzyme activities and DLS mass (i.e. average measures) Mass distributions of complexes (e.g. SEC and GEMMA) Mass distributions of complexes (e.g. SEC and GEMMA) Validate omic-predicted interactions and model them Validate omic-predicted interactions and model them PEPCC (Harry Gill): Systematically express, purify and characterize interaction suspects (e.g. of core wnt signaling proteins) PEPCC (Harry Gill): Systematically express, purify and characterize interaction suspects (e.g. of core wnt signaling proteins)
 Enzyme activities and DLS mass (i.e. average measures) Mass distributions of complexes (e.g. SEC and GEMMA) Mass distributions of complexes (e.g. SEC and GEMMA) Validate omic-predicted interactions and model them Validate omic-predicted interactions and model them PEPCC (Harry Gill): Systematically express, purify and characterize interaction suspects (e.g. of core wnt signaling proteins) PEPCC (Harry Gill): Systematically express, purify and characterize interaction suspects (e.g. of core wnt signaling proteins).")
4
Ultimate Goal Present Future Safer flying airplanes with autopilots Safer flying airplanes with autopilots Ultimate Goal: individualized, state feedback based clinical trials Ultimate Goal: individualized, state feedback based clinical trials Better understanding => better control Better understanding => better control Conceptual models help trial designs today Conceptual models help trial designs today Computer models of airplanes help train pilots and autopilots Computer models of airplanes help train pilots and autopilots Radivoyevitch et al. (2006) BMC Cancer 6:104
 BMC Cancer 6:104.")
5
dNTP Supply System Figure 1. dNTP supply. Many anticancer agents act on or through this system to kill cells. The most central enzyme of this system is RNR. UDP CDP GDP ADP dTTP dCTP dGTP dATP dT dC dG dA DNA dUMP dU TS DCTD dCK DNA polymerase TK1 cytosol mitochondria dT dC dG dA TK2 dGK dTMP dCMP dGMP dAMP dTTP dCTP dGTP dATP 5NT NT2 cytosol nucleus dUDP dUTP dUTPase dN dCK flux activation inhibition ATP or dATP RNR dCK

6
R1 R2 R1 R2 UDP, CDP, GDP, ADP bind to catalytic site ATP, dATP, dTTP, dGTP bind to selectivity site dATP inhibits at activity site, ATP activates at activity site? 5 catalytic site states x 5 s-site states x 3 a-site states x 2 h-site states = 150 states (150) 6 different hexamer complexes => 2^(150) 6 models ~ 1 followed by a trillion zeros ATP activates at hexamerization site?? Ribonucleotide Reductase (RNR) R2 RNR is Combinatorially Complex
 6 different hexamer complexes => 2^(150) 6 models ~ 1 followed by a trillion zeros ATP activates at hexamerization site . Ribonucleotide Reductase (RNR) R2 RNR is Combinatorially Complex.")
7
Michaelis-Menten Model RNR: no NDP and no R2 dimer => k cat of complex is zero, else different R1-R2-NDP complexes can have different k cat values. E + S ES

8
solid line = Eqs. (1-2) dotted = Eq. (3) Data from Scott, C. P., Kashlan, O. B., Lear, J. D., and Cooperman, B. S. (2001) Biochemistry 40(6), 1651-166 R=R1 r=R2 2 G=GDP t=dTTP Substitute this in here to get a quadratic in [S] which solves as Bigger systems of higher polynomials cannot be solved algebraically => use ODEs (above) Michaelis-Menten Model [S] vs. [S T ] [S] vs. [S T ] (3)
 Biochemistry 40(6), R=R1 r=R2 2 G=GDP t=dTTP Substitute this in here to get a quadratic in [S] which solves as Bigger systems of higher polynomials cannot be solved algebraically => use ODEs (above) Michaelis-Menten Model [S] vs. [S T ] [S] vs. [S T ] (3).")
9
E ES EI ESI E ES EI ESI E ES EI E ES EI ESI E EI ESI E ES ESI E EI ESI E ES ESI = = E EI E ESI E ES E Competitive inhibition uncompetitive inhibition if k cat_ESI =0 E | ES EI | ESI noncompetitive inhibition Example of K=K’ Model = = Enzyme, Substrate and Inhibitor

10
Total number of spur graph models is 16+4=20 Radivoyevitch, (2008) BMC Systems Biology 2:15 Rt Spur Graph Models R RR RRtt RRt Rt R RRtt RRt Rt R RR RRtt Rt R RR RRt Rt R RR RRtt RRt R RRtt Rt R RRt Rt R RR Rt IJJJ JJIJ JJJIJIJJ IJIJIJJIJJII JJJJ R RRtt RRt R RR RRtt R RR RRt R Rt R RRtt R RRt JIIJ IIJJ JIJI R RR R IJII IIIJIIJI JIII IIII R Rt R RRtt R RRt R RR I0II III0 II0I0III R = R1 t = dTTP for dTTP induced R1 dimerization (RR, Rt, RRt, RRtt)
 BMC Systems Biology 2:15 Rt Spur Graph Models R RR RRtt RRt Rt R RRtt RRt Rt R RR RRtt Rt R RR RRt Rt R RR RRtt RRt R RRtt Rt R RRt Rt R RR Rt IJJJ JJIJ JJJIJIJJ IJIJIJJIJJII JJJJ R RRtt RRt R RR RRtt R RR RRt R Rt R RRtt R RRt JIIJ IIJJ JIJI R RR R IJII IIIJIIJI JIII IIII R Rt R RRtt R RRt R RR I0II III0 II0I0III R = R1 t = dTTP for dTTP induced R1 dimerization (RR, Rt, RRt, RRtt)")
11
R t RR t Rt R t RRt t Rt RRtt K d_R_R K d_Rt_R K d_Rt_Rt K d_R_t K d_RRt_t K d_RR_t = = = = | | | Rt Grid Graph Models R t RR t Rt R t RRt t RRtt K R_R K R_t K RRt_t K RR_t = = = = = = = = = = = = = = = = = = = R t RR t Rt R t RRt t Rt RRtt K R_R K Rt_R K Rt_Rt K R_t | | | | | | | | | | | | | | | ? ? HIFF HDFFHDDD =

12
AIC c = N*log(SSE/N)+2P+2P(P+1)/(N-P-1) Scott, C. P., Kashlan, O. B., Lear, J. D., and Cooperman, B. S. (2001) Biochemistry 40(6), 1651-166 Radivoyevitch, (2008) BMC Systems Biology 2:15 Application to Data HDFF = = R RRtt IIIJ III0m ModelParameterInitial Value Optimal Value Confidence Interval 1 III0mm190.00082.368(79.838, 84.775) SSE4397.550525.178 AIC71.96557.090 2 IIIJR2t21.000^32.725^3(2.014^3, 3.682^3) SSE2290.516557.797 AIC67.39957.512 27 HDFFR2t01.00012369.79(0, 1308627507869) R1t0_t1.0001.744(0.003, 1187.969) R2t0_t1.0000.010(0.000, 403.429) SSE25768.23477.484 AIC105.34277.423
 Biochemistry 40(6), Radivoyevitch, (2008) BMC Systems Biology 2:15 Application to Data HDFF = = R RRtt IIIJ III0m ModelParameterInitial Value Optimal Value Confidence Interval 1 III0mm (79.838, ) SSE AIC IIIJR2t21.000^32.725^3(2.014^3, 3.682^3) SSE AIC HDFFR2t (0, ) R1t0_t (0.003, ) R2t0_t (0.000, ) SSE AIC")
13
2+5+9+13 = 28 parameters => 2 28 =2.5x10 8 spur graph models via K j =∞ hypotheses 28 models with 1 parameter, 428 models with 2, 3278 models with 3, 20475 with 4 R = R1 X = ATP Yeast R1 structure. Dealwis Lab, PNAS 102, 4022-4027, 2006 ATP-induced R1 Hexamerization Kashlan et al. Biochemistry 2002 41:462

15
Kolmogorov-Smirnov Test p < 10 -16 28 of top 30 did not include an h-site term; 28/30 ≠ 503/2081 with p < 10 -16 This suggests no h-site. Top 13 included R6X8 or R6X9, save one, single edge model R6X7 This suggests that less than 3 a-sites are occupied in hexamer. 2088 Models with SSE < 2 min (SSE) Data from Kashlan et al. Biochemistry 2002 41:462
 Data from Kashlan et al. Biochemistry :462.")
16
Conclusions (so far) 1.The dataset does not support the existence of an h-site 2.The dataset suggests that ~1/2 of the a-site are not occupied by ATP R1 a a [ATP]=~1000[dATP] So system prefers to have 3 a-sites empty and ready for dATP Inhibition versus activation is partly due to differences in pockets a a a a UDP CDP GDP ADP dTTP dCTP dGTP dATP dT dC dG dA DNA dUMP dU TS DCTD dCK DNA polymerase TK1 cytosol mitochondria dT dC dG dA TK2 dGK dTMP dCMP dGMP dAMP dTTP dCTP dGTP dATP 5NT NT2 cytosol nucleus dUDP dUTP dUTPase dN dCK flux activation inhibition ATP or dATP RNR dCK
![Conclusions (so far) 1.The dataset does not support the existence of an h-site 2.The dataset suggests that ~1/2 of the a-site are not occupied by ATP R1 a a [ATP]=~1000[dATP] So system prefers to have 3 a-sites empty and ready for dATP Inhibition versus activation is partly due to differences in pockets a a a a UDP CDP GDP ADP dTTP dCTP dGTP dATP dT dC dG dA DNA dUMP dU TS DCTD dCK DNA polymerase TK1 cytosol mitochondria dT dC dG dA TK2 dGK dTMP dCMP dGMP dAMP dTTP dCTP dGTP dATP 5NT NT2 cytosol nucleus dUDP dUTP dUTPase dN dCK flux activation inhibition ATP or dATP RNR dCK](http://images.slideplayer.com/25/8038000/slides/slide_16.jpg "Conclusions (so far) 1.The dataset does not support the existence of an h-site 2.The dataset suggests that ~1/2 of the a-site are not occupied by ATP R1 a a [ATP]=~1000[dATP] So system prefers to have 3 a-sites empty and ready for dATP Inhibition versus activation is partly due to differences in pockets a a a a UDP CDP GDP ADP dTTP dCTP dGTP dATP dT dC dG dA DNA dUMP dU TS DCTD dCK DNA polymerase TK1 cytosol mitochondria dT dC dG dA TK2 dGK dTMP dCMP dGMP dAMP dTTP dCTP dGTP dATP 5NT NT2 cytosol nucleus dUDP dUTP dUTPase dN dCK flux activation inhibition ATP or dATP RNR dCK")
17
X 8 K Models x 8 k Models => 64 Models Tetrameric Enzyme Models (e.g. Thymidine Kinase 1) If [S] ≈ [S T ]
 If [S] ≈ [S T ].")
18
Complete K vs. Binary K

19
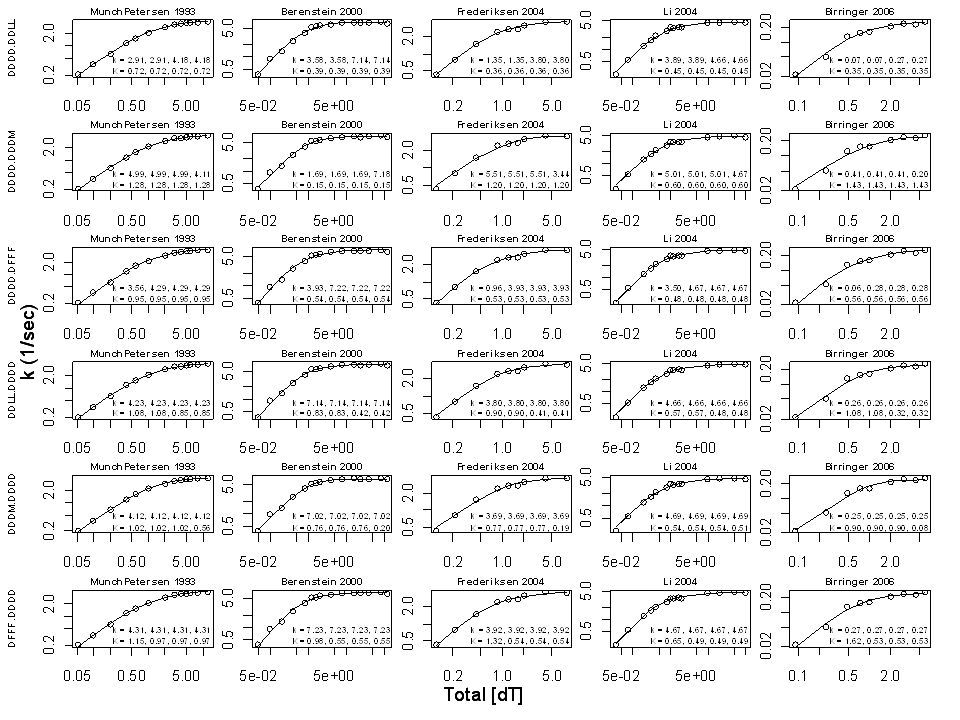
K = (0.85, 0.69, 0.65, 0.51) µM k = (3.3, 3.9, 4.1, 4.1) sec -1 k max = 4.1/sec S 50 = 0.6 µM h = 1.1 DFFF.DDDD DDLL.DDDD DDDM.DDDD DDDD.DFFF DDDD.DDLL DDDD.DDDM
 µM k = (3.3, 3.9, 4.1, 4.1) sec -1 k max = 4.1/sec S 50 = 0.6 µM h = 1.1 DFFF.DDDD DDLL.DDDD DDDM.DDDD DDDD.DFFF DDDD.DDLL DDDD.DDDM")
21
8-parameter model fits to the datasets

22
K = (10 -6, 10 -3, 1, 10 3 ) A) k = (100, 10, 40, 10) B) k = (10, 10, 40, 100) With parameters initially at ½ their true values, A identified itself, B had difficulties (its 1 st two K estimates were off 2-5-fold). Horizontal lines are k 1 /4, 2k 2 /4, 3k 3 /4 and 4k 4 /4 Vertical lines are K 1 /4, 2K 2 /3, 3K 3 /2 and 4K 4

23
Combinatorially Complex Equilibrium Model Selection (ccems, CRAN 2009) Systems Biology Markup Language interface to R (SBMLR, BIOC 2004) Model networks of enzymes Model individual enzymes SUMMARY R1 R2 R1 R2
 Systems Biology Markup Language interface to R (SBMLR, BIOC 2004) Model networks of enzymes Model individual enzymes SUMMARY R1 R2 R1 R2")
24
Figure 8. T. Thorsen et al. (S. R. Quake Lab) Science 2002 Figure 9. J. Melin and S. R. Quake Annu. Rev. Biophys. Biomol. Struct. 2007. 36:213–31 Background: Quake Lab Microfluidics Figure 9 shows how a peristaltic pump is implemented by three valves that cycle through the control codes 101, 100, 110, 010, 011, 001, where 0 and 1 represent open and closed valves; note that the 0 in this sequence is forced to the right as the sequence progresses.

25
Adaptive Experimental Designs Find best next 10 measurement conditions given models of data collected. Need automated analyses in feedback loop of automatic controls of microfluidic chips

26
5 0 10 + - setpoint KpKp KiKi ∫ Σ hot plate water temperature Simple example of a control system for a single-input single-output (SISO) system
 system")
27
Emphasis is on the stochastic component of the model. Is there something in the black box or are the input wires disconnected from the output wires such that only thermal noise is being measured? Do we have enough data? Model components: (Deterministic = signal) + (Stochastic = noise) Statistics Engineering Emphasis is on the deterministic component of the model We already know what is in the box, since we built it. The goal is to understand it well enough to be able to control it. Predict the best multi-agent drug dose time course schedules Increasing amounts of data/knowledge Why Systems Biology
 + (Stochastic = noise) Statistics Engineering Emphasis is on the deterministic component of the model We already know what is in the box, since we built it. The goal is to understand it well enough to be able to control it. Predict the best multi-agent drug dose time course schedules Increasing amounts of data/knowledge Why Systems Biology.")
28
dNTPs + Analogs DNA + Drug-DNA Damage Driven or S-phase Driven dNTP demand is either DNA repair Salvage De novo MMR - Cancer Treatment Strategy IUdR

29
Conclusions For systems biology to succeed: For systems biology to succeed: –move biological research toward systems which are best understood –specialize modelers to become experts in biological literatures (SB is not a service)
")
30
Acknowledgements Case Comprehensive Cancer Center Case Comprehensive Cancer Center NIH (K25 CA104791) NIH (K25 CA104791) Chris Dealwis (CWRU Pharmacology) Chris Dealwis (CWRU Pharmacology) Anders Hofer (Umea) Anders Hofer (Umea) Thank you Thank you
 NIH (K25 CA104791) Chris Dealwis (CWRU Pharmacology) Chris Dealwis (CWRU Pharmacology) Anders Hofer (Umea) Anders Hofer (Umea) Thank you Thank you")
31
Indirect Approach pro-B Cell Childhood ALL T: TEL-AML1 with HR T: TEL-AML1 with HR t : TEL-AML1 with CCR t : TEL-AML1 with CCR t : other outcome t : other outcome B: BCR-ABL with CCR B: BCR-ABL with CCR b: BCR-ABL with HR b: BCR-ABL with HR b: censored, missing, or other outcome b: censored, missing, or other outcome Ross et al: Blood 2003, 102:2951-2959 Yeoh et al: Cancer Cell 2002, 1:133-143 Radivoyevitch et al., BMC Cancer 6, 104 (2006)
")
32
THF CH 2 THF CH 3 THF CHOTHF DHF CHODHF HCHO GAR FGAR AICAR FAICAR dUMP dTMP NADP + NADPH NADP + NADPH NADP + NADPH MetHcys Ser Gly GART ATIC TS ATP ADP 11R 2R 2 3 4 10 9 8 5 6 7 12 11 13 HCOOH MTHFD MTHFR MTR DHFR SHMT FTS FDS Morrison PF, Allegra CJ: Folate cycle kinetics in human breast cancer cells. JBiolChem 1989, 264:10552-10566.

33
library(ccems) # Thymidine Kinase Example topology = list( heads=c("E1S0"), # E1S0 = substrate free E sites=list( c=list( # c for catalytic site t=c("E1S1","E1S2","E1S3","E1S4") ) # t for tetramer ) ) # TK1 is 25kDa = 25mg/umole, so 1 mg =.04 umoles g = mkg(topology, activity=T,TCC=F) dd=subset(TK1,(year==2000),select=c(E,S,v)) dd=transform(dd, ET=ET/4,v=ET*v/(.04*60))# now uM/sec tops=ems(dd,g,maxTotalPs=8,kIC=10,topN=96)#9 min on 1 cpu library(ccems) # Ribonucleotide Reductase Example topology <- list( heads=c("R1X0","R2X2","R4X4","R6X6"), sites=list( # s-sites are already filled only in (j>1)-mers a=list( #a-site thread m=c("R1X1"), # monomer 1 d=c("R2X3","R2X4"), # dimer 2 t=c("R4X5","R4X6","R4X7","R4X8"), # tetramer 3 h=c("R6X7","R6X8","R6X9","R6X10", "R6X11", "R6X12") # hexamer 4 ), # tails of a-site threads are heads of h-site threads h=list( # h-site m=c("R1X2"), # monomer 5 d=c("R2X5", "R2X6"), # dimer 6 t=c("R4X9", "R4X10","R4X11", "R4X12"), # tetramer 7 h=c("R6X13", "R6X14", "R6X15","R6X16", "R6X17", "R6X18")# hexamer 8 ) g=mkg(topology,TCC=TRUE) dd=subset(RNR,(year==2002)&(fg==1)&(X>0),select=c(R,X,m,year)) cpusPerHost=c("localhost" = 4,"compute-0-0"=4,"compute-0-1"=4,"compute-0-2"=4) top10=ems(dd,g,cpusPerHost=cpusPerHost, maxTotalPs=3, ptype="SOCK",KIC=100) K = e ∆G/RT => K = (0, ∞) maps to ∆G = (-∞, ∞) Model weights e ∆AIC /∑e ∆AIC
 # Thymidine Kinase Example topology = list( heads=c( E1S0 ), # E1S0 = substrate free E sites=list( c=list( # c for catalytic site t=c( E1S1 , E1S2 , E1S3 , E1S4 ) ) # t for tetramer ) ) # TK1 is 25kDa = 25mg/umole, so 1 mg =.04 umoles g = mkg(topology, activity=T,TCC=F) dd=subset(TK1,(year==2000),select=c(E,S,v)) dd=transform(dd, ET=ET/4,v=ET*v/(.04*60))# now uM/sec tops=ems(dd,g,maxTotalPs=8,kIC=10,topN=96)#9 min on 1 cpu library(ccems) # Ribonucleotide Reductase Example topology <- list( heads=c( R1X0 , R2X2 , R4X4 , R6X6 ), sites=list( # s-sites are already filled only in (j>1)-mers a=list( #a-site thread m=c( R1X1 ), # monomer 1 d=c( R2X3 , R2X4 ), # dimer 2 t=c( R4X5 , R4X6 , R4X7 , R4X8 ), # tetramer 3 h=c( R6X7 , R6X8 , R6X9 , R6X10 , R6X11 , R6X12 ) # hexamer 4 ), # tails of a-site threads are heads of h-site threads h=list( # h-site m=c( R1X2 ), # monomer 5 d=c( R2X5 , R2X6 ), # dimer 6 t=c( R4X9 , R4X10 , R4X11 , R4X12 ), # tetramer 7 h=c( R6X13 , R6X14 , R6X15 , R6X16 , R6X17 , R6X18 )# hexamer 8 ) g=mkg(topology,TCC=TRUE) dd=subset(RNR,(year==2002)&(fg==1)&(X>0),select=c(R,X,m,year)) cpusPerHost=c( localhost = 4, compute-0-0 =4, compute-0-1 =4, compute-0-2 =4) top10=ems(dd,g,cpusPerHost=cpusPerHost, maxTotalPs=3, ptype= SOCK ,KIC=100) K = e ∆G/RT => K = (0, ∞) maps to ∆G = (-∞, ∞) Model weights e ∆AIC /∑e ∆AIC")
34
Comments on Methods

35
Conjecture Greater X/R ratio dominates at high Ligand concentrations In this limit the system wants to partition As much ATP into a bound form as possible Bias in 1 st four residuals => Do not weight errors

Similar presentations










 Bruck Caltech joint work with Brian Fett.>")




 Involvement.>")




