Download presentation
Presentation is loading. Please wait.

1
The Basic Local Alignment Search Tool (BLAST) Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs
 Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs")
2
The Basic Local Alignment Search Tool (BLAST) A Y W T Y I V A L T – Q V R Q Y E A T S I L C I V M I Y S R A - Q Y R Y W R Y Most local alignments contain highly conserved sections without gaps
 A Y W T Y I V A L T – Q V R Q Y E A T S I L C I V M I Y S R A - Q Y R Y W R Y Most local alignments contain highly conserved sections without gaps")
3
The Basic Local Alignment Search Tool (BLAST) A Y W T Y I V A L T – Q V R Q Y E A T S I L C I V M I Y S R A - Q Y R Y W R Y -> search for high scoring segment pairs (HSP), i.e. gap-free local alignments

4
The Basic Local Alignment Search Tool (BLAST)
")
5
A Y W T Y I V A L T – Q V R Q Y E A T S I L C I V M I Y S R A - Q Y R Y W R Y Advantages: (a) speed (b) statistical theory about HSP exists.
 speed (b) statistical theory about HSP exists.")
6
The Basic Local Alignment Search Tool (BLAST) Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs (2) Use word pairs as seeds
 Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs (2) Use word pairs as seeds")
7
Pair-wise sequence alignment T W L M H C A Q Y I C I M X H X C X T H Y (1) Search word pairs of length 3 with score > T, Use them as seeds.
 Search word pairs of length 3 with score > T, Use them as seeds.")
8
Pair-wise sequence alignment Naïve algorithm would have a complexity of O(l 1 * l 2 ) Solution: Preprocess query sequence: Compile a list of all words that have a Score > T when aligned to a word in the Query.
 Solution: Preprocess query sequence: Compile a list of all words that have a Score > T when aligned to a word in the Query.")
9
Pair-wise sequence alignment Naïve algorithm would have a complexity of O(l 1 * l 2 ) Solution: Preprocess query sequence: Compile a list of all words that have a Score > T when aligned to a word in the Query. Complexity: O(l 1 ) Organize words in efficient data structure (tree) for fast look-up
 Organize words in efficient data structure (tree) for fast look-up.")
10
The Basic Local Alignment Search Tool (BLAST) Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs (2) Use word pairs as seeds (3) Extend seed alignments until score drops below threshold value
 Rapid data base search tool (1990) Idea: (1) Search for high scoring segment pairs (2) Use word pairs as seeds (3) Extend seed alignments until score drops below threshold value")
11
Pair-wise sequence alignment T W L M H C A Q Y I C I M X H X C X T H Y Extend seeds until score drops by X.

12
Pair-wise sequence alignment T W L M H C A Q Y I C I X M X H X C X T X H X Y Extend seeds until score drops by X.

13
Pair-wise sequence alignment Algorithm not guaranteed to find best segment pair (Heuristic) But works well in practice!
 But works well in practice!")
14
The Basic Local Alignment Search Tool (BLAST) New BLAST version (1997) Two-hit strategy
 New BLAST version (1997) Two-hit strategy")
15
Pair-wise sequence alignment W L M H C A Q Y A R V I M X H X C X T H W A X R X v X Search two word pairs of at the same diagonal, use lower threshold T

16
The Basic Local Alignment Search Tool (BLAST) New BLAST version (1997) Two-hit strategy Gapped BLAST Position-Specific Iterative BLAST (PSI BLAST)
 New BLAST version (1997) Two-hit strategy Gapped BLAST Position-Specific Iterative BLAST (PSI BLAST)")
17
The Basic Local Alignment Search Tool (BLAST)
")
18
Multiple sequence alignment 1aboA 1.NLFVALYDfvasgdntlsitkGEKLRVLgynhn..............gE 1ycsB 1 kGVIYALWDyepqnddelpmkeGDCMTIIhrede............deiE 1pht 1 gYQYRALYDykkereedidlhlGDILTVNkgslvalgfsdgqearpeeiG 1ihvA 1.NFRVYYRDsrd......pvwkGPAKLLWkg.................eG 1vie 1.drvrkksga.........awqGQIVGWYctnlt.............peG 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

19
Multiple sequence alignment First question: how to score multiple alignments? Possible scoring scheme: Sum-of-pairs score

20
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

21
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

22
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP......

23
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQtkngqGWVPSNYITPVN 1ycsB 39 WWWARlndkeGYVPRNLLGLYP

24
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

25
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

26
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp

27
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp

28
Multiple sequence alignment Multiple alignment implies pairwise alignments: 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

29
Multiple sequence alignment Multiple alignment implies pairwise alignments: Use sum of scores of these p.a. 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN......

30
Multiple sequence alignment Goal: Find multi-alignment with maximum score !

31
Multiple sequence alignment Needleman-Wunsch coring scheme can be generalized from pair-wise to multiple alignment Multidimensional search space instead of two- dimensional matrix!

32
Multiple sequence alignment

33
Complexity: For sequences of length l 1 * l 2 * l 3 O( l 1 * l 2 * l 3 ) For n sequences ( average length l ): O( l n ) Exponential complexity!
 For n sequences ( average length l ): O( l n ) Exponential complexity!")
34
Multiple sequence alignment Needleman-Wunsch coring scheme can be generalized from pair-wise to multiple alignment Optimal solution not feasible:

35
Multiple sequence alignment Needleman-Wunsch coring scheme can be generalized from pair-wise to multiple alignment Optimal solution not feasible: -> Heuristics necessary

36
Multiple sequence alignment (A) Carillo and Lipman (MSA) Find sub-space in dynamic-programming Matrix where optimal path can be found
 Carillo and Lipman (MSA) Find sub-space in dynamic-programming Matrix where optimal path can be found")
37
Multiple sequence alignment (B) Stoye, Dress (DCA) Divide search space into small Calculate optimal alignment for sub-spaces Concatenate sub-alignments
 Stoye, Dress (DCA) Divide search space into small Calculate optimal alignment for sub-spaces Concatenate sub-alignments")
38
Multiple sequence alignment (B) Stoye, Dress (DCA)
 Stoye, Dress (DCA)")
39
Multiple sequence alignment (B) Stoye, Dress (DCA)
 Stoye, Dress (DCA)")
40
Multiple sequence alignment Progressive alignment. Carry out a series of pair-wise alignment

41
Most popular way of constructing multiple alignments: Progressive alignment. Carry out a series of pair-wise alignment Multiple sequence alignment

42
WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP Multiple sequence alignment

43
WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP Align most similar sequences Multiple sequence alignment

44
WCEAQTKNGQGWVPSNYITPVN WW--RLNDKEGYVPRNLLGLYP- AVVIQDNSDIKVVP--KAKIIRD YAVESEASFQPVAALERIN WLNYNEERGDFPGTYVEYIGRKKISP

45
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN WW--RLNDKEGYVPRNLLGLYP- AVVIQDNSDIKVVP--KAKIIRD YAVESEASVQ--PVAALERIN------ WLN-YNEERGDFPGTYVEYIGRKKISP

46
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN WW--RLNDKEGYVPRNLLGLYP- AVVIQDNSDIKVVP--KAKIIRD YAVESEASVQ--PVAALERIN------ WLN-YNEERGDFPGTYVEYIGRKKISP Align sequence to alignment

47
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN- WW--RLNDKEGYVPRNLLGLYP- AVVIQDNSDIKVVP--KAKIIRD YAVESEASVQ--PVAALERIN------ WLN-YNEERGDFPGTYVEYIGRKKISP Align alignment to alignment

48
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN-------- WW--RLNDKEGYVPRNLLGLYP-------- AVVIQDNSDIKVVP--KAKIIRD------- YAVESEA---SVQ--PVAALERIN------ WLN-YNE---ERGDFPGTYVEYIGRKKISP

49
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN-------- WW--RLNDKEGYVPRNLLGLYP-------- AVVIQDNSDIKVVP--KAKIIRD------- YAVESEA---SVQ--PVAALERIN------ WLN-YNE---ERGDFPGTYVEYIGRKKISP Rule: “once a gap - always a gap”

50
Multiple sequence alignment Order of pair-wise profile alignments determined by phylogenetic tree based on pair-wise similarity values (guide tree)
")
51
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP

52
Multiple sequence alignment WCEAQTKNGQGWVPSNYITPVN WWRLNDKEGYVPRNLLGLYP AVVIQDNSDIKVVPKAKIIRD YAVESEAHPGSFQPVAALERIN WLNYNETTGERGDFPGTYVEYIGRKKISP

53
Multiple sequence alignment Problem: simple guide tree determines multiple alignment; multiple alignment determines phyolgeneitc analysis

54
Multiple sequence alignment Implementations: Clustal W, PileUp, MultAlin

55
Local multiple alignment M M

56
M M M

57
M M M M´

58
Local multiple alignment Find motifs contained in all sequences in data set Problem: motifs often present in only sub-families

59
Neither local nor global methods appliccable

60
Alignment possible if order conserved

61
The DIALIGN approach

62
Combination of local and global methods.

63
The DIALIGN approach Combination of local and global methods. Find local pair-wise similarities between input sequences (fragments)
.")
64
The DIALIGN approach Combination of local and global methods. Find local pair-wise similarities between input sequences (fragments) Compose alignments from fragments
 Compose alignments from fragments.")
65
The DIALIGN approach Combination of local and global methods. Find local pair-wise similarities between input sequences (fragments) Compose alignments from fragments Ignore non-related parts of the sequences
 Compose alignments from fragments Ignore non-related parts of the sequences.")
66
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc

67
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc

68
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc

69
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc

70
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc ------atctaatagttaaaccccctcgtgcttag-------agatccaaac cagtgcgtgtattactaac----------ggttcaatcgcgcacatccgc--

71
The DIALIGN approach atctaatagttaaactcccccgtgcttagagatccaaac cagtgcgtgtattactaacggttcaatcgcgcacatccgc ------atctaatagttaaaccccctcgtgcttag-------agatccaaac cagtgcgtgtattactaac----------ggttcaatcgcgcacatccgc-- ------atcTAATAGTTAaaccccctcgtGCTTag-------AGATCCaaac cagtgcgtgTATTACTAAc----------GGTTcaatcgcgcACATCCgc--

72
The DIALIGN approach Score of an alignment: Define score of fragment f: l(f) = length of f s(f) = sum of matches (similarity values) P(f) = probability to find a fragment with length l(f) and at least s(f) matches in random sequences that have the same length as the input sequences. Score w(f) = -ln P(f)
 = -ln P(f).")
73
The DIALIGN approach Score of an alignment: Define score of alignment as sum of scores w(f) of its fragments No gap penalty is used! Optimization problem for pair-wise alignment: Find chain of fragments with maximal total score

74
The DIALIGN approach ------atctaatagttaaaccccctcgtgcttag-------agatccaaac cagtgcgtgtattactaac----------ggttcaatcgcgcacatccgc-- Fragment-chaining algorithm finds optimal chain of fragments.

75
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

76
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

77
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

78
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

79
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

80
The DIALIGN approach Multiple fragment alignment atctaatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

81
The DIALIGN approach Multiple fragment alignment atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaagagtatcacccctgaattgaataa

82
The DIALIGN approach Multiple fragment alignment atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaacggttcaatcgcg caaa--gagtatcacccctgaattgaataa

83
The DIALIGN approach Multiple fragment alignment atc------taatagttaaactcccccgtgcttag cagtgcgtgtattactaac----------ggttcaatcgcg caaa--gagtatcacc----------cctgaattgaataa

84
The DIALIGN approach Multiple fragment alignment atc------taatagttaaactcccccgtgc-ttag cagtgcgtgtattactaac----------gg-ttcaatcgcg caaa--gagtatcacc----------cctgaattgaataa

85
The DIALIGN approach Multiple fragment alignment atc------taatagttaaactcccccgtgc-ttag cagtgcgtgtattactaac----------gg-ttcaatcgcg caaa--gagtatcacc----------cctgaattgaataa Consistency: it is possible to introduce gaps such that all segment pairs are aligned.

86
The DIALIGN approach Multiple fragment alignment atc------TAATAGTTAaactccccCGTGC-TTag cagtgcGTGTATTACTAAc----------GG-TTCAATcgcg caaa--GAGTATCAcc----------CCTGaaTTGAATaa

87
Program evaluation Use biologically verified alignments (known 3D structure of proteins) Compare alignments produced by computer programs to “biologically correct” alignments.
 Compare alignments produced by computer programs to biologically correct alignments.")
88
Program evaluation (1) First evaluation of multiple alignment programs (McClure, Vasi, Fitch,1994) 4 protein families used: Globin, kinase, protease, ribonuclease H, all globally related -> global programs performed best
 First evaluation of multiple alignment programs (McClure, Vasi, Fitch,1994) 4 protein families used: Globin, kinase, protease, ribonuclease H, all globally related -> global programs performed best")
89
Program evaluation (2) The BAliBASE (Thompson et al., 1999) ~ 100 protein families with known 3D structure, some with large insertions/deletions.
 The BAliBASE (Thompson et al., 1999) ~ 100 protein families with known 3D structure, some with large insertions/deletions.")
90
Program evaluation 1aboA 1.NLFVALYDfvasgdntlsitkGEKLRVLgynhn..............gE 1ycsB 1 kGVIYALWDyepqnddelpmkeGDCMTIIhrede............deiE 1pht 1 gYQYRALYDykkereedidlhlGDILTVNkgslvalgfsdgqearpeeiG 1ihvA 1.NFRVYYRDsrd......pvwkGPAKLLWkg.................eG 1vie 1.drvrkksga.........awqGQIVGWYctnlt.............peG 1aboA 36 WCEAQt..kngqGWVPSNYITPVN...... 1ycsB 39 WWWARl..ndkeGYVPRNLLGLYP...... 1pht 51 WLNGYnettgerGDFPGTYVEYIGrkkisp 1ihvA 27 AVVIQd..nsdiKVVPRRKAKIIRd..... 1vie 28 YAVESeahpgsvQIYPVAALERIN...... Key alpha helix RED beta strand GREEN core blocks UNDERSCORE

91
Program evaluation Results: Four programs performed best, but no method was best in all test examples. ClustalW, SAGA and RPPR best for global alignment, DIALIGN best for sequences with large insertions or deletions.

92
Program evaluation (3) Lassmann and Sonnhammer (2002) Used BAliBASE plus artificial sequences for local alignment Results: T-COFFEE best for closely related sequences, DIALIGN best for distal sequences.
 Lassmann and Sonnhammer (2002) Used BAliBASE plus artificial sequences for local alignment Results: T-COFFEE best for closely related sequences, DIALIGN best for distal sequences.")
93
Program evaluation

94
Alignment of large genomic sequences Important tool for identifying functional sites (e.g. genes or regulatory elements)
.")
95
Alignment of large genomic sequences Phylogenetic Footprinting: Functional sites more conserved during evolution => Sequence similarity indicates biological function

96
Alignment of large genomic sequences DIALIGN performs well in identifying local homologies, but is slow

97
Quadratic program running time

104
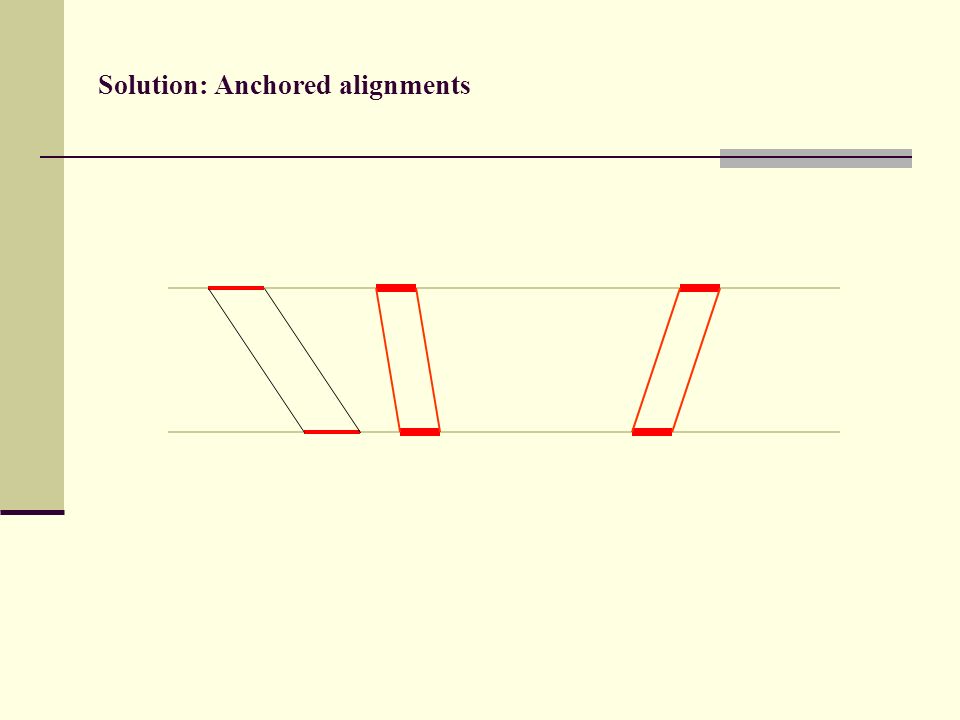
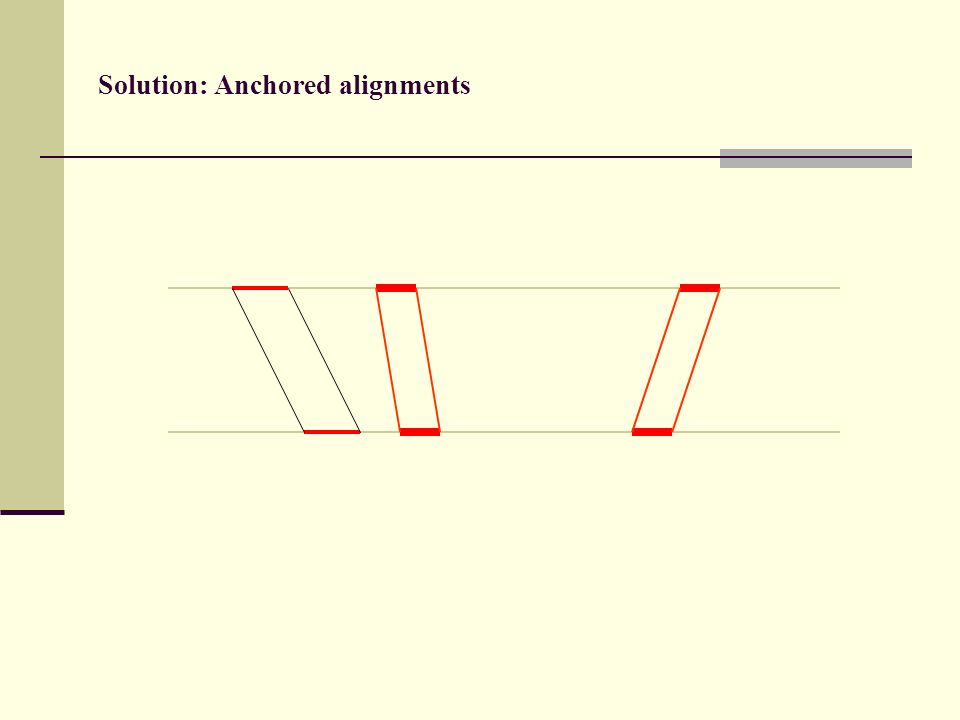
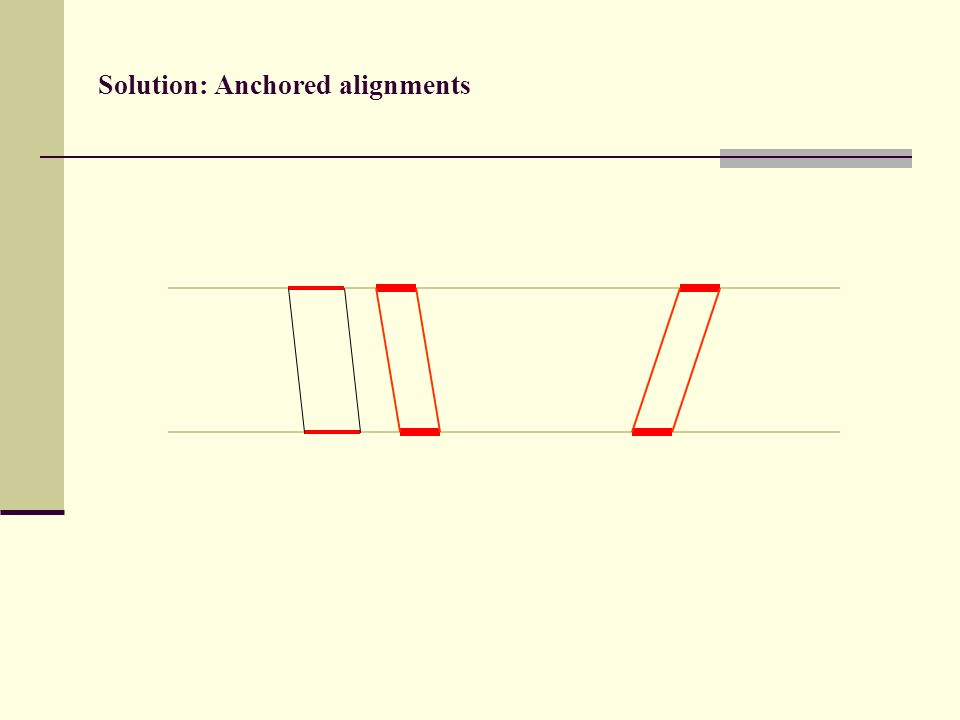
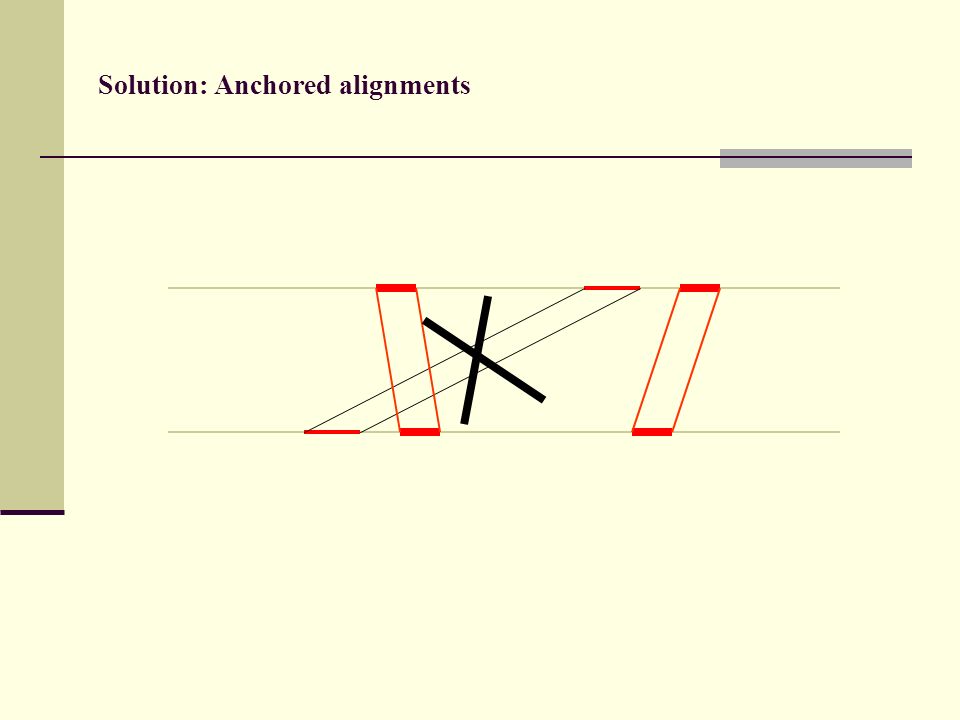
Solution: Anchored alignments

111
Find anchor points to reduce search space

112
Solution: Anchored alignments Use fast heuristic method to find anchor points: CHAOS developed together with Mike Brudno Brudno et al. (2003), BMC Bioinformatics 4:66
, BMC Bioinformatics 4:66.")
113
Solution: Anchored alignments

114
(3) Anchored alignments
 Anchored alignments")
116
First step to gene prediction: Exon discovery by genomic alignment

117
Evaluation of different alignment programs: Compare local sequence similarity identified by alignment programs to known exons Morgenstern et al. (2002), Bioinformatics 18:777-787
, Bioinformatics 18:")
118
DIALIGN alignment of human and murine genomic sequences

119
DIALIGN alignment of tomato and Thaliana genomic sequences

120
Evaluation of DIALIGN, PipMaker, WABA, BLASTN and TBLASTX on a set of 42 human and murine genomic sequences. Compare similarities to annotated exons Apply cut-off parameter to resulting alignments Measure sensitivity and specificity

121
Performance of long-range alignment programs for exon discovery (human - mouse comparison)
")
122
Performance of long-range alignment programs for exon discovery (thaliana - tomato comparison)
")
123
AGenDA: Alignment-based Gene Detection Algorithm Bridge small gaps between DIALIGN fragments -> cluster of fragments Search conserved splice sites and start/stop codons at cluster boundaries to Identify candidate exons Recursive algorithm finds biologically consistent chain of potential exons

124
Identification of candidate exons Fragments in DIALIGN alignment

125
Identification of candidate exons Build cluster of fragments

126
Identification of candidate exons Identify conserved splice sites

127
Identification of candidate exons Candidate exons bounded by conserved splice sites

128
Construct gene models using candidate exons Score of candidate exon (E) based on DIALIGN scores for fragments, score of splice junctions and penalty for shortening / extending Find biologically consistent chain of candidate exons (starting with start codon, ending with stop codon, no internal stop codons …) with maximal total score
 based on DIALIGN scores for fragments, score of splice junctions and penalty for shortening / extending Find biologically consistent chain of candidate exons (starting with start codon, ending with stop codon, no internal stop codons …) with maximal total score")
129
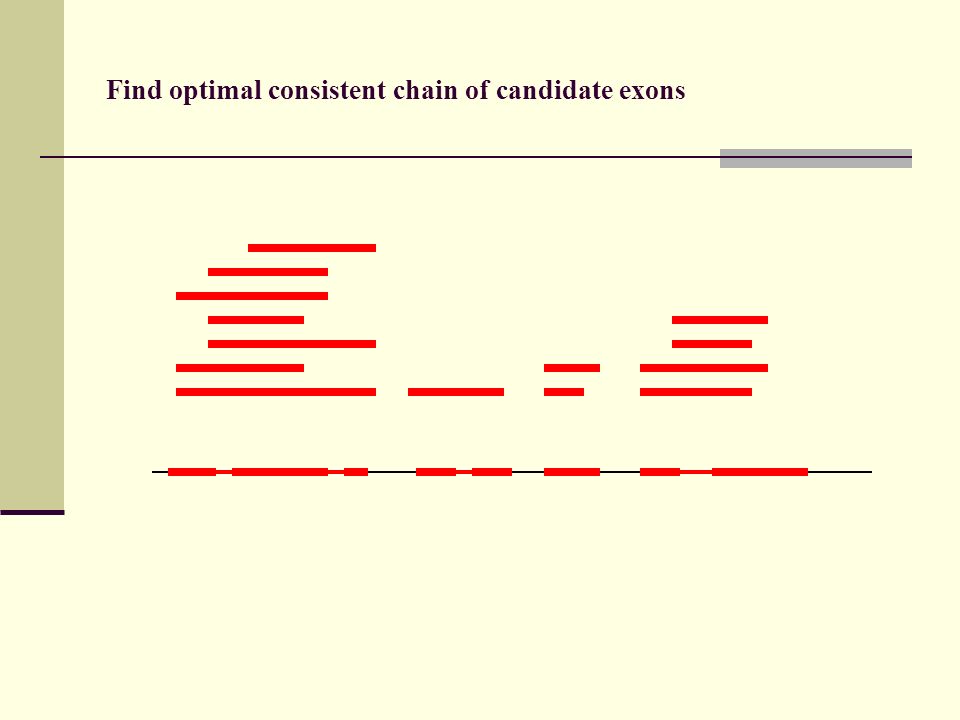
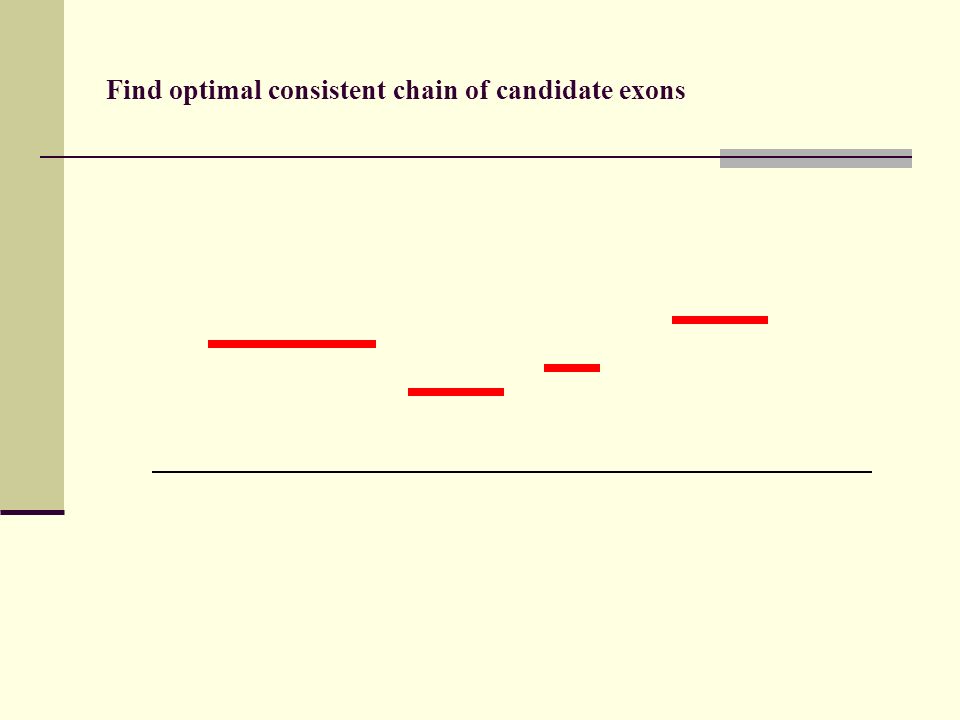
Find optimal consistent chain of candidate exons

133
atggtaggtagtgaatgtga

134
Find optimal consistent chain of candidate exons atggtaggtagtgaatgtga G1G2

135
Find optimal consistent chain of candidate exons Recursive algorithm calculates optimal chain of candidate exons in N log N time

136
DIALIGN fragments

137
Candidate exons

138
Complete model

139
Results: 105 pairs of genomic sequences from human and mouse (Batzoglou et al., 2000)
")
140
AGenDA GenScan 64 % 12 % 17 %

141
Results: Quality of AGenDA-based gene models comparable to results from GenScan Exons identified that have not been identified by GenScan No statistical models derived from known genes (no training data necessary!) Method generally appliccable
 Method generally appliccable")
142
AGenDA: Alignment-based Gene Detection Algorithm WWW server: http://bibiserv/TechFak.Uni-Bielefeld.DE/agenda Rinner, Taher, Goel, Sczyrba, Brudno, Batzoglou, Morgenstern, submitted

Similar presentations



















>")


 useful for alignment of genomic sequences. Possible applications: Detection.>")



