Download presentation
Presentation is loading. Please wait.

1
Genetic Mutations SDK October 8, 2013

2
OBJECTIVES By the end of this session the student should be able to:
Define Mutation Frequency of mutations in normal individuals Classify different types of mutation Explain the mechanism of mutation Explain the role of mutation in biodiversity Explain how mutations can cause severe diseases Give examples of deletions, duplications, and insertions in genes Define trinucleotide repeat expansions and how they cause neurological diseases SDK 2012

3
What is a gene mutation? Replacement or change of a nucleotide base with another, in one or both strands, or addition or deletion of a base pair in a DNA molecule . Mutations are changes in genetic material(Nitrogenous bases) – changes in DNA code – thus a change in a gene(s) In gene mutations, the DNA code will have a base (or more) missing, added, or exchanged in a codon. SDK 2012
 – changes in DNA code – thus a change in a gene(s) In gene mutations, the DNA code will have a base (or more) missing, added, or exchanged in a codon. SDK")
4
Gene mutation out come Mutations can lead to missing or malformed proteins, and that can lead to disease. SDK 2012

5
Types of Mutations Germ-line mutations .Mutations that are inherited from parents are called germ-line mutations. Acquired mutations. Mutations that are acquired during your lifetime are called acquired mutations Some mutations happen during cell division, when DNA gets duplicated. Still other mutations are caused when DNA gets damaged by environmental factors, including UV radiation, chemicals, and viruses. SDK 2012

6
How common are mutations?
Mutations occurs at a frequency of about 1 in every 1 billion base pairs Everybody has about 5-10 potentially deadly mutations in our genes- in each cell of our body! SDK 2012

7
When everyone has mutations, Why they are not always seen
Diseases caused by just one copy of a defective gene are not manifested with the exception of Huntington's disease, which is rare and afflicted carriers are more likely to die before reproducing. Most inherited genetic diseases are recessive, which means that a person must inherit two copies of the mutated gene to inherit a disorder. This is one reason that marriage between close relatives is discouraged; two genetically similar adults are more likely to give a child two copies of a defective gene. They are not always seen because the mutation may have occurred in a section of DNA that doesn’t make a protein. SDK 2012

8
Mutations Outcome The affected gene may still function.
Mutations may be harmful. Mutations may be beneficial. Mutations may have no effect on the organism. Mutations are a major source of genetic variation in a population increasing biodiversity. SDK 2012

9
Mutations a cause of Biodiversity
SDK 2012

10
Does all mutations passed on to next generation?
NO Only mutations in gametes (egg & sperm) are passed onto offspring(Germline Mutation). Mutations in somatic cells (body cells) only affect the body in which they occur and are not passed onto offspring. SDK 2012
 are passed onto offspring(Germline Mutation). Mutations in somatic cells (body cells) only affect the body in which they occur and are not passed onto offspring. SDK")
11
TRUE Brain Work 1 A mutation may happen in any gene. TRUE OR FALSE?
SDK 2012

12
Spontaneous and Induced Mutations
Spontaneous: Occur spontaneously without obvious reason. Induced mutations: caused by mutagens. Mutagens are the agent that causes the DNA code to change (mutate) X-Ray, Chemicals, UV light, Radiation, etc SDK 2012
 X-Ray, Chemicals, UV light, Radiation, etc. SDK")
13
UV Light (Sun Light) Brain Work 2
Which of the following may cause mutations? Coffee UV light (sun light) Hair gel Vaccines UV Light (Sun Light) SDK 2012
 Hair gel. Vaccines. UV Light (Sun Light) SDK")
14
Original The fat cat ate the wee rat.
Types of Mutations Point mutations. A point mutation is a simple change in one base of the gene sequence. 2. Frame shift mutations. one or more bases are inserted or deleted Original The fat cat ate the wee rat. Point Mutation The fat cat ate the wet rat. Original The fat cat ate the wee rat Frame Shift The fat caa tet hew eer at SDK 2012

15
Morphological Types of Point mutations
Transitions. Transitions occur when a Purine is converted to a purine (A to G or G to A) Pyrimide is converted to a pyrimidine (T to C or C to T) 2. Transversion. A transversion results when Purine is converted to a pyrimidine (A to C or G to T) Pyrimidine is converted to a purine. (T to A or C to G) SDK 2012
 Pyrimide is converted to a pyrimidine (T to C or C to T) 2. Transversion. A transversion results when. Purine is converted to a pyrimidine (A to C or G to T) Pyrimidine is converted to a purine. (T to A or C to G) SDK")
16
Types of Mutations according to their effects on the protein (or mRNA).
Silent Mutations. Mutation in a codons that produce same amino acid. These mutations affect the DNA but not the protein. Therefore they have no effect on the organism’s phenotype. CUU CUC Missense Mutations. Missense mutations substitute one amino acid for another. Example. HbS, Sickle Cell Hemoglobin, is a change in the beta-globin gene, where a GAG codon is converted to GUG GAG GUG Nonsense mutations. convert an amino acid into a stop codon. The effect is to shorten the resulting protein. Sometimes this has only a little effect, however, often nonsense mutations result in completely non-functional proteins. UUU UAA\ UGA SDK 2012

17
SDK 2012

18
Frame-shift In a frameshift mutation one or more bases are inserted, or deleted. Because our cells read our DNA in three letter words, adding or removing one letter changes each subsequent word. This type of mutation can make the DNA sequence meaningless . For example: Original= THE FAT CAT ATE THE WEE RAT FRAMESHIFT= THE FAT CAA TET HEW EER AT. SDK 2012

19
Brain Work 3 Mutations are a natural part of the cellular process reproduction. The cell has tools that catch and repair 99.9% of mutations. TRUE OR FALSE? TRUE SDK 2012

20
TRUE Brain Work 4 Most mutations are caught and repaired in the cell.
TRUE or FALSE? TRUE SDK 2012

21
Classical Types of Point mutation Mutations
Point mutation occurs when the base sequence of a codon is changed. (ex. GCA is changed to GAA) There are 3 types: Substitution Deletion Also called frameshift mutations Insertion SDK 2012
 There are 3 types: Substitution. Deletion. Also called frameshift mutations. Insertion. SDK")
22
Substitution A substitution is a mutation that exchanges one base for another (i.e. a change in a single “chemical letter” such as switching an A to G. For example: CTGGAG CTGGGG SDK 2012

23
Substitution Mutations
Normal DNA: CGA – TGC – ATC Alanine – Threonine - stop Mutated DNA: CGA – TGC – TTC Alanine – Threonine - Lysine This is a substitution mutation What will happen to the amino acids? What has happened to the DNA? The adenine was replaced with thymine SDK 2012

24
The cat ate the rat The hat ate the rat SDK 2012

25
THE CAT ATE THE RAT Analogy
3 letter words because codons are 3 letters THE CAT ATE THE RAT SUBSTITUTION Thc cat ate the rat. May have little effect. You still have the idea like a typo on a test. The hat ate the rat. Changes the thought of the sentence. The effect Depends on where the substitution happens SDK 2012

26
Clinical Examples SDK 2012

27
Sickle Cell Anemia Sickle cell anemia is the result of a (substitution) point mutation in codon 6 of the -globin gene resulting in the substitution of amino acid glutamic acid by valine SDK 2012
 point mutation in codon 6 of the -globin gene resulting in the substitution of amino acid glutamic acid by valine. SDK")
28
Sickle Cell Anemia Under conditions of low oxygen tension, such as
Following exercise or In an atmosphere containing a low oxygen level, The following changes occur: The haemoglobin agglutinates to form insoluble rod-shaped polymers Red blood cells become distorted and sickle-shaped The sickle-shaped cells rupture easily causing haemolytic anaemia The sickle shaped cells tend to block capillaries interfering with the blood flow to various organs. SDK 2012

29
Thalassaemia Substitution of C by U in mRNA that is coding globin chain of 146 amino acid. Resulted in the formation of a stop signal UAG in place CAG of glutamate in codon number 39. This result in a shortened globin chain containing only 39 instead of the normal 146 amino acids in the -globin protein chain. This protein is functionally useless and is equivalent to absence of -globin gives clinical symptoms of thalassaemia, SDK 2012

30
Thalassaemia SDK 2012

31
Thalassemias Beta thalassaemia is a genetic disorder in which there is lack of beta globin. It may be the result of: Deletion of the whole gene so that beta globin cannot not produced (designated o ) Deletion of the promoter region so that transcription cannot occur (designated o ) Deletion of a large part of the gene resulting in a grossly abnormal or reduced synthesis functional protein (designated + ) SDK 2012
 Deletion of the promoter region so that transcription cannot occur (designated o ) Deletion of a large part of the gene resulting in a grossly abnormal or reduced synthesis functional protein (designated + ) SDK")
32
Clinical Features of -Thalassaemia
Haemoglobin A (α2 2) cannot be produced Hb F (α2 g2) is produced even in adults Hb A2 (α 2 d2) formation is increased Eerythrocytes are microcytic (small) due to lack of normal haemoglobin Erythrocytes rupture easily causing severe haemolytic anaemia, requiring repeated blood transfusions The bone marrow expands trying to compensate by increasing haemopoiesis. SDK 2012
 cannot be produced. Hb F (α2 g2) is produced even in adults. Hb A2 (α 2 d2) formation is increased. Eerythrocytes are microcytic (small) due to lack of normal haemoglobin. Erythrocytes rupture easily causing severe haemolytic anaemia, requiring repeated blood transfusions. The bone marrow expands trying to compensate by increasing haemopoiesis. SDK")
33
Clinical Features of -Thalassaemia
The bones of the face and skull are thickened causing a characteristic facial appearance The spleen and liver enlarge because haemopoietic tissue forms in them Excess iron accumulates in the blood and is deposited in the heart, liver, pancreas and other organs (this is because of repeated transfusions while no blood is actually lost from the body) Children have delayed growth and development and are prone to repeated infections SDK 2012
 Children have delayed growth and development and are prone to repeated infections. SDK")
34
Point Mutation In alpha-Globin Gene “Elongated α Globin Chain, Haemoglobin Constant spring \Wayne Hb” Here the stop codon UAA at position 142 in the alpha (-) globin gene was substituted by the codon for glutamine. Translation of the protein thus continued until a stop codon was encountered at codon 173. The -globin was considerably elongated, resulting in a variant of haemoglobin termed Haemoglobin Constant spring\ Wayne Hb. SDK 2012
 globin gene was substituted by the codon for glutamine. Translation of the protein thus continued until a stop codon was encountered at codon 173. The -globin was considerably elongated, resulting in a variant of haemoglobin termed Haemoglobin Constant spring\ Wayne Hb. SDK")
35
Elongated α Globin Chain Or Haemoglobin Wayne
Hemoglobin Constant Spring. Hemoglobin Constant Spring is a variant in which a mutation in the alpha globin gene produces an alpha globin chain that is abnormally long. The quantity of hemoglobin in the cells is low for two reasons. First, the messenger RNA for hemoglobin Constant Spring is unstable. Some is degraded prior to protein synthesis. Second, the Constant Spring alpha chain protein is itself unstable. The result is a thalassemic phenotype. (The designation Constant Spring derives from the isolation of the hemoglobin variant in a family of ethnic Chinese background from the Constant Spring district of Jamaica.) SDK 2012
 SDK")
36
CTGGAG CTGGCCTAG Insertion
Insertions are mutations in which extra base pairs are inserted into a new place in the DNA. CTGGAG CTGGCCTAG SDK 2012

37
Insertion Mutations What will happen to the amino acids?
Normal DNA: CGA – TGC – ATC Alanine – Threonine – stop Mutated DNA: CGA – TAG – CAT – C Alanine – Isoleucine – Valine An adenine was inserted thereby pushing all the other bases over a frame. This is an insertion mutation, also a type of frameshift mutation. What has happened to the DNA? What will happen to the amino acids? SDK 2012

38
Insertion Mutations The cat ate the rat The cca tat eth era t SDK 2012

39
Analogy Insertion The cat ate the rat. The cca tat eth era t.
Inserting the c causes a FRAMESHIFT THE SENTENCE NO LONGER MAKES SENSE!! Insertions may have huge effects. SDK 2012

40
Haemophilia A An X-linked recessive disorder in which blood clotting does not occur due to deficiency of clotting factor VIII. In most cases the mutation is the result of insertion of a large segment, consisting of about 3800 bp, in the coding region of the factor VIII. This results in total inactivation of the protein. SDK 2012

41
Haemophilia A Inherited as a sex linked recessive trait with bleeding manifestations only in males. Genes which control factor VIII and IX production are located on the x chromosome Affected male marries a normal female: none of sons will be affected, all daughters will be carriers Female carrier marries normal male: 50% chance sons will be affected and 50% chance daughters will be carriers SDK 2012

42
Deletion Deletions involve removal of one or more base pairs. They vary greatly in size from deletion of a single base to deletion of a whole gene. The clinical effects often depend on the size and location of the deleted part of the gene. CTGGAG CT AG SDK 2012

43
Deletion Mutations What has happened to the DNA?
Normal DNA: CGA – TGC – ATC Alanine – Threonine – stop Mutated DNA: CGA – TCA- TC Alanine – Serine What has happened to the DNA? A guanine was deleted, thereby pushing all the bases down a frame. What will happen to the amino acids? This is called a deletion mutation, also a type of frameshift mutation. SDK 2012

44
The sentence no longer makes sense!! Deletions can have huge effects.
Analogy DELETION The cat ate the rat. Thc ata tet her at FRAMESHIFT The sentence no longer makes sense!! Deletions can have huge effects. SDK 2012

45
Muscular Dystrophy Deletions of Dystrophin Gene
Dystrophin is a protein that is an important component of skeletal muscle. The dystrophin gene is located on the p arm of the X chromosome (Xp21.2). It is a very large gene spanning 2.5 million bp of genomic DNA and consists of 79 exons coding for a protein of approximately 3600 amino acids (11kb). The gene for dystrophin production sits on the X chromosome. If a normal gene for dystrophin is present, then the protein will be made. If the gene is missing or altered, dystrophin may not be produced at all or only in abnormal forms, resulting in Duchenne muscular dystrophy SDK 2012
. It is a very large gene spanning 2.5 million bp of genomic DNA and consists of 79 exons coding for a protein of approximately 3600 amino acids (11kb). The gene for dystrophin production sits on the X chromosome. If a normal gene for dystrophin is present, then the protein will be made. If the gene is missing or altered, dystrophin may not be produced at all or only in abnormal forms, resulting in Duchenne muscular dystrophy. SDK")
46
SDK 2012

47
SDK 2012

48
Muscular Dystrophy Deletions of Dystrophin Gene
Deletion of the whole or most of the dystrophin gene Dystrophin may not be produced at all Or produce in in abnormal forms, Resulting in Duchenne muscular dystrophy This is a severe X-linked recessive disorder that affects boys and is transmitted by carrier females. X linked Recessive disorder. In affected boys there is almost complete lack of dystrophin, muscle weakness beginning in childhood and increasing progressively in severity so that the individual is wheel-chair bound at the age of about 15 years. Death usually ensues in the early twenties due to respiratory muscle involvement. SDK 2012

49
X linked Recessive disorder
SDK 2012

50
Becker Muscular Dystrophy(BMD)
Deletions involving a small non-critical part of the gene result in altered dystrophin. This causes the clinical condition of Becker muscular dystrophy in BMD muscle weakness begins in adolescence and is very slowly progressive, and affected individuals may lead an almost normal life. SDK 2012

51
Cystic Fibrosis. Cystic fibrosis (CF) is a genetic condition that affects many organs in the body: especially the lungs, pancreas and sweat glands. Cystic fibrosis is caused by a mutation in the Cystic Fibrosis Trans-membrane Regulator (CFTR) gene, that is located on chromosome 7. This gene Produces a trans-membrane protein that regulates the flow of chloride ions into the cells. The most common mutation is termed the ∆508 mutation, which is a deletion of a single codon at position number 508 in exon 10 of the CFTR gene. Homozygotes(both parents need to be the carriers of the defective gene) for a ∆508 mutation have cystic fibrosis disease.
 gene, that is located on chromosome 7. This gene Produces a trans-membrane protein that regulates the flow of chloride ions into the cells. The most common mutation is termed the ∆508 mutation, which is a deletion of a single codon at position number 508 in exon 10 of the CFTR gene. Homozygotes(both parents need to be the carriers of the defective gene) for a ∆508 mutation have cystic fibrosis disease.")
52
Gene That Encodes CFTR The gene that encodes the CFTR protein is found on the human chromosome 7, on the long arm at position q31.2. Mutations consist of replacements, duplications, deletions or shortenings in the CFTR gene. This may result in proteins that may not function, work less effectively, are more quickly degraded, or are present in inadequate numbers SDK 2012

53
Cystic Fibrosis. The defective gene produces a defective protein leading to a blockage in the transportation of the salt, thus leading to production of thick, sticky mucus. SDK 2012

54
Presentation The secretion of very thick, sticky mucus causes
Obstruction of the bronchi and predisposing to pulmonary infections, Pancreatic duct obstruction leads to problems with digestion. Intestinal and liver problems and When it blocks the sweat glands, it leads to loss of excessive salt through sweat. This leads to imbalance of minerals within the body. SDK 2012

55
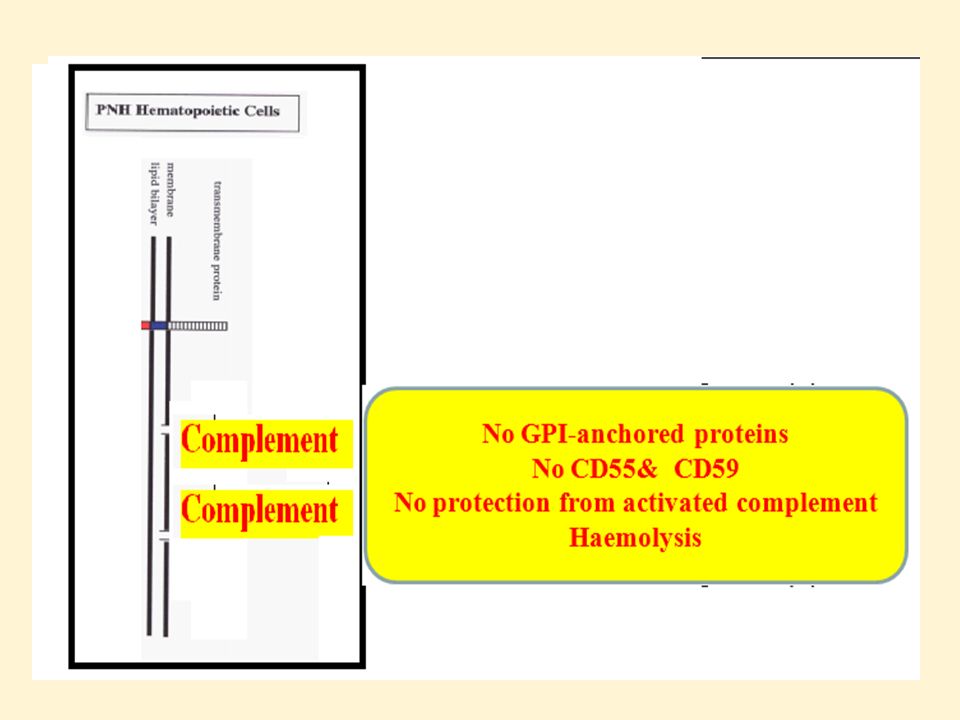
Paroxysmal Nocturnal Hemoglobinurea
Paroxysmal nocturnal hemoglobinuria is a disorder of blood cells in which absence of specific molecule(GPI anchor protein, CD55 [Decay Accelerating Factor (DAF)], and CD59 [Membrane Inhibitor of Complement Lysis (MIRL)] on the surface of the cells (particularly RBC) leads to premature destruction of the cells by the complement system. This destruction is intermittent (paroxysmal). SDK 2012
], and CD59 [Membrane Inhibitor of Complement Lysis (MIRL)] on the surface of the cells (particularly RBC) leads to premature destruction of the cells by the complement system. This destruction is intermittent (paroxysmal). SDK")
56
Paroxysmal Nocturnal Hemoglobinurea
GPI anchor protein on the surface of red blood cells produced by the bone marrow stem cells This is caused by a mutation of PIG-A gene, PIG-A gene is present on the X chromosome important in making GPI protein anchors. Defect makes the red cells in susceptible to destruction by the complement system.

57
Paroxysmal Nocturnal Hemoglobinurea
The PIG-A mutation occurs in a bone marrow stem cell. All the blood cells made by this defective stem cell are deficient in GPI-anchored proteins. (glycosyl-phosphatidylinositol GPI). The GPI-anchored proteins are present on the surface of red blood cells that protect red cells from the activity of the complement system. When they are absent , no protection from complement and this lead to intravascular haemolysis.
. The GPI-anchored proteins are present on the surface of red blood cells that protect red cells from the activity of the complement system. When they are absent , no protection from complement and this lead to intravascular haemolysis.")
58
Genetics PIGA gene(phosphatidylinositol glycan class A) is present in in the X chromosome and can have several mutations, from deletions to point mutations. The genetic mutation leading to the inability to synthesize the glycosyl-phosphatidylinositol (GPI) anchor proteine.
 is present in in the X chromosome and can have several mutations, from deletions to point mutations. The genetic mutation leading to the inability to synthesize the glycosyl-phosphatidylinositol (GPI) anchor proteine.")
60
Complement Activation Lysed PNH RBCs and free hemoglobin in the plasma
RBC Lysis Normal RBCs PNH RBC CD59 Complement Activation Chronic Hemolysis Schematic of the effect of terminal complement activation on normal RBCs and PNH RBCs The absence of the complement inhibitor CD59 renders the PNH RBCs susceptible to hemolysis Chronic hemolysis results in anemia, poor quality of life, and an increased risk for thrombotic events Intact RBC Lysed PNH RBCs and free hemoglobin in the plasma

61
Role of CD55 &CD 59

62
Paroxysmal Nocturnal Hemoglobinurea
The term "nocturnal" refers to the belief that hemolysis is triggered by acidosis during sleep and activates complement to hemolyze an unprotected and abnormal RBC membrane. However, this observation was later disproved. Hemolysis has been shown to occur throughout the day and is not actually paroxysmal, but the urine concentrated overnight produces the dramatic change in color. The essential group of membrane proteins that are lacking in all hematopoietic cells are called complement-regulating surface proteins, including the decay-accelerating factor (DAF), or CD55; homologous restriction factor (HRF), or C8 binding protein; and membrane inhibitor of reactive lysis (MIRL), or CD59. All of these proteins interact with complement proteins, particularly C3b and C4b, dissociate the convertase complexes of the classic and alternative pathways, and halt the amplification of the activation process. Hemolytic anemia is due to intravascular destruction of RBCs by complement, with varying degrees in the absence of these regulating proteins resulting in uncontrolled amplification of the complement system that leads to destruction of the RBC membrane.
, or CD55; homologous restriction factor (HRF), or C8 binding protein; and membrane inhibitor of reactive lysis (MIRL), or CD59. All of these proteins interact with complement proteins, particularly C3b and C4b, dissociate the convertase complexes of the classic and alternative pathways, and halt the amplification of the activation process. Hemolytic anemia is due to intravascular destruction of RBCs by complement, with varying degrees in the absence of these regulating proteins resulting in uncontrolled amplification of the complement system that leads to destruction of the RBC membrane.")
63
The codons 92 to 97 of the -globin gene are deleted.
Deletion of 6 codons in the -globin gene resulting in a variant Hemoglobin The codons 92 to 97 of the -globin gene are deleted. This results in a shortened -globin protein that produces a haemoglobin variant termed Haemoglobin Gun Hill. In homozygotes it produces mild clinical; symptoms. SDK 2012

64
Codon no Codon Amino acid 91 CUG Leucine 92 CAC Histidine 93 UGU
Cysteine 94 GAC Aspartic acid 95 AAG Lysine 96 97 SDK 2012

65
Frame shift mutations Frame shift mutations involve a deletion or insertion of one or two base pairs within a coding sequence of a gene. As the coding message is read in triplets codons and deletions will altered the the reading frame of mRNA This results in a non-sense sequence of amino acids till stop codon. Original= THE FAT CAT ATE THE WEE RAT Frame-shift= THE FAT CAA TET HEW EER AT SDK 2012

66
Frame Shift Mutations(Deletion)
An example occurs in the -globin gene in which one nucleotide of codon 39 is deleted leads to altered sequence. SDK 2012

67
Frame Shift Mutations(Insertion)
Insertion of a sequence of bases into a coding sequence of a gene. Sometimes a whole gene sequence may be duplicated. Hereditary motor and sensory neuropathy type I A DNA segment at locus 17p11 is duplicated. Tay-Sachs Disease SDK 2012

68
Tay-Sachs Disease Tay-sachs disease is an autosomal recessive disorder
This genetic defect is located in the HEXA (hexosaminidase) gene, which is found on chromosome 15. The hexa gene makes part of an enzyme called beta-hexosaminidase A This enzyme helps break down a fatty substance called GM2 ganglioside in nerve cells. Mutations in the HEXA gene disrupt the activity of beta-hexosaminidase A, preventing the breakdown of the fatty substances. As a result, the fatty substances accumulate to deadly levels in the brain and spinal cord. The buildup of GM2 ganglioside causes progressive damage to the nerve cells. SDK 2012
 gene, which is found on chromosome 15. The hexa gene makes part of an enzyme called beta-hexosaminidase A. This enzyme helps break down a fatty substance called GM2 ganglioside in nerve cells. Mutations in the HEXA gene disrupt the activity of beta-hexosaminidase A, preventing the breakdown of the fatty substances. As a result, the fatty substances accumulate to deadly levels in the brain and spinal cord. The buildup of GM2 ganglioside causes progressive damage to the nerve cells. SDK")
69
Tay-Sachs Disease SDK 2012

70
Tay-Sachs Disease SDK 2012

71
Tay-Sachs Disease SDK 2012

72
Tay-Sachs Disease S\S Loss of hearing Physical and mental retardation
Seizures Dementia And most noticeably detected by the red dots it causes on the retina of an individuals eye SDK 2012

73
Trinucleotide Repeat Expansions
Trinucleotides are triplets of nucleotides that are repeated The number of repeats varies in different individuals. Trinucleotide repeats is CAG CAG CAG CAG CAG Trinucleotide repeats are widespread in the genome, and may occur in exons, introns, promoter sequences or non-coding regions. They are perfectly normal and occur in all individuals However a mutation arises when the repeats become unstable and undergo expansion, namely an increase in the number of repeats as they are transmitted from one generation to the next. When the number of repeats exceeds a certain limit, clinical symptoms occur. An example is the huntingtin gene, which, when mutated, causes Huntington's disease. Normal range of (CAG)n is 11 to 34 Huntington's disease appear in individuals in whom the number of repeats is greater than 37. SDK 2012
n is 11 to 34. Huntington s disease appear in individuals in whom the number of repeats is greater than 37. SDK")
74
Huntington's Disease Autosomal Dominant Inheritance
Due to an excess of C-A-G nucleotide repeats(>37) in the HTT gene on the short arm of chromosome 4 forms (4p 16.3)Huntington's protein. Huntington's protein has increase number of glutamine (polyglutamine). Excess of repeats causes the protein to form aggregates that are deposited within the neurons causing neuronal degeneration Affects brain and spinal cord, especially the basal ganglia. SDK 2012
 in the HTT gene on the short arm of chromosome 4 forms (4p 16.3)Huntington s protein. Huntington s protein has increase number of glutamine (polyglutamine). Excess of repeats causes the protein to form aggregates that are deposited within the neurons causing neuronal degeneration. Affects brain and spinal cord, especially the basal ganglia. SDK")
75
Trinucleotide Repeat Expansions
SDK 2012

76
Clinical Manifestations
Most commonly appear in mid-forties If appear at a younger age, more severe Manifestations occur because of wasting away of brain cells. Sudden jerky, involuntary movements throughout body Difficulties with balance and coordination Dysphasia Hesitant/slurred speech Progressive dysfunction of intellectual and thought processes (dementia) Cognitive deficits a. Working memory loss b. Reduced capacity to plan, organize, and sequence 7. Restlessness, irritability 8. Depression or Euphoria SDK 2012
 Cognitive deficits. a. Working memory loss. b. Reduced capacity to plan, organize, and sequence. 7. Restlessness, irritability. 8. Depression or Euphoria. SDK")
77
What happens to a person who has a mutation?
Most of the time the mutation is harmless because 95% sections of DNA do not code for protein (junk DNA). But sometimes the mutations can cause disorders such as Huntington’s disease and sickle cell anemia etc. SDK 2012
. But sometimes the mutations can cause disorders such as Huntington’s disease and sickle cell anemia etc. SDK")
78
Flyaway Mutation Brain Work 5
Which of these is NOT a type of mutation. a) Point mutation b) Flyaway mutation c) Frameshift mutation d) Nonsense mutation Flyaway Mutation SDK 2012
 Point mutation. b) Flyaway mutation. c) Frameshift mutation. d) Nonsense mutation. Flyaway Mutation. SDK")
79
Brain Work 6 The correct answer is B deletion.
1. THE CAT SAW THE FAT RAT 2. THE CAT SAW THE RAT The change in Statement 1 to form Statement 2 is most similar to what type of mutation? Insertion Deletion Substitution Frameshift The correct answer is B deletion. Because the sentence is missing the word fat which occurs in deletion as it is the removal of a section of DNA. SDK 2012

80
Brain Work 7 5′AGAUCGAGU3′ → 5’ACAUCGAGU3′
The chain above represents three codons Which of the following changes would be expected in the amino acid chain if the mutation shown above occurred? The amino acid sequence would be shorter than expected. The identity of one amino acid would change. The amino acid sequence would remain unchanged. The identities of more than one amino acid would change. The correct answer is B because according to the codon chart if G is switched by a C then one amino acid is affected because now instead of coding for Arginine it now codes for Threonine. SDK 2012

81
Thank You SDK 2012

Similar presentations


>")
 Mutations.>")












 Mutations. What is a gene mutation? Parts of DNA will have a base (or more) missing, added, or incorrect A mistake in the genetic code Wrong.>")


