Download presentation
Presentation is loading. Please wait.

2
Background The disease first appeared in the medical literature when endocrinologists Prader, Labhart, and Willi published a report describing an unusual pattern of abnormalities. Because of this report, the disease got its name, Prader-Willi Syndrome.

3
Introduction What is Prader-Willi Syndrome? PWS is characterized by diminished fetal activity, obesity, muscular hypotonia, mental retardation, short stature, hypogonadotropic hypogonadism, and small hands and feet.

4
Symptom Behavioral problems, usually during transitions and unanticipated changes, such as stubbornness or temper tantrums Delayed motor skills and speech Cognitive problems, ranging from near normal intelligence to mild mental retardation; learning disabilities are common Low sex hormone levels

5
The Genetic Changes Related to PWS Most cases of PWS (about 70 percent) occur when the segment 11-13 of the paternal chromosome 15 is deleted in each cell.
 occur when the segment of the paternal chromosome 15 is deleted in each cell.")
6
The region is called Prader-Willi syndrome chromosome region (PWCR). It includes the following genes: SNRPN gene SNRPN gene is also known as SM-D. The protein encoded by this gene is one polypeptide of a small nuclear ribonucleoprotein complex.

7
The protein plays a role in pre-mRNA processing, possibly tissue-specific alternative splicing events. The 5' UTR of this gene has been identified as an imprinting center. Alternative splicing or deletion caused by a translocation event in this paternally-expressed region is responsible for Prader-Willi switch failure. syndrome due to parental imprint

8
Necdin gene This intronless gene is located in the Prader- Willi syndrome deletion region. It is an imprinted gene and is expressed exclusively from the paternal allele. The protein coded by the gene maintains the dTMP pool critical for DNA replication and repair. Studies in mouse suggest that the protein may suppress growth in postmitotic neurons.

9
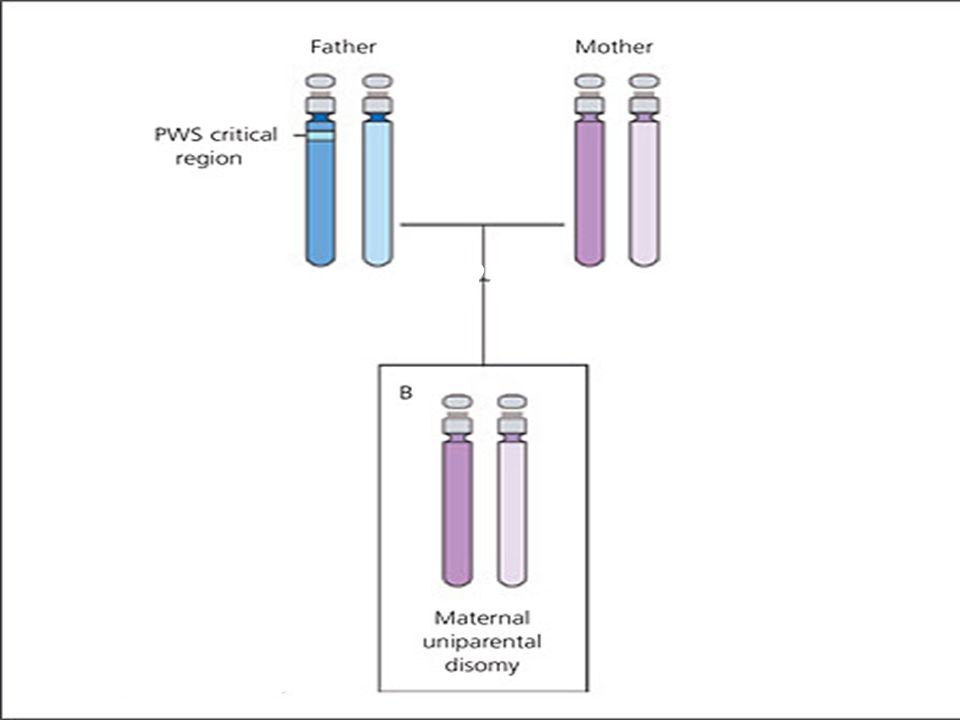
Continue In another 25 percent of cases, a person with PWS has two copies of chromosome 15 inherited from his or her mother instead of one copy from each parent. This phenomenon is called maternal uniparental disomy.. Rarely, PWS can also be caused by a chromosomal rearrangement called a translocation, or by a mutation or other defect that abnormally turns off genes on the paternal chromosome 15.

10
Stages of Development Infancy Babies with PWS are very floppy at birth, and the ability to suck is weak or absent. Childhood Some time between the ages of one and four years, children with PWS begin to show a heightened interest in food and in severe cases develop what appears to be an insatiable appetite, so that they will try to obtain food by any means possible.

11
Adolescence People with PWS do not usually reach full sexual development, and there have been only three cases worldwide of a woman with PWS having a child. However there may be cases of which doctors are unaware. Life as an adult As adults, people with PWS have varying abilities in attaining independence, although all will need some form of support or monitoring to help with controlling their food intake, and thus their weight.

12
The probability of inheritance of PWS Most cases of PWS are not inherited, particularly those caused by a deletion in the paternal chromosome 15 or by maternal uniparental disomy. These genetic changes occur as random events during the formation of reproductive cells (eggs and sperm) or in early embryonic development. Affected people typically have no history of the disorder in their family. Rarely, a genetic change responsible for PWS can be inherited. For example, it is possible for a genetic defect that abnormally inactivates genes on the paternal chromosome 15 to be passed from one generation to the next.
 or in early embryonic development. Affected people typically have no history of the disorder in their family. Rarely, a genetic change responsible for PWS can be inherited. For example, it is possible for a genetic defect that abnormally inactivates genes on the paternal chromosome 15 to be passed from one generation to the next..")
13
Diagnosis A suspected diagnosis of PWS is usually made by a physician based on clinical symptoms. The diagnosis is then confirmed by a blood test. Two types of tests can be used to confirm the diagnosis. A "FISH" (fluorescent in situ hybridization) test will identify those patients with PWS due to a deletion, but will not identify those who have PWS by "UPD" (uniparental disomy) or via an imprinting error.
 test will identify those patients with PWS due to a deletion, but will not identify those who have PWS by UPD (uniparental disomy) or via an imprinting error..")
14
Continue Methylation analysis will detect all three forms of PW, so if PWS is suspected but a FISH test is negative, a DNA methylation test is warranted. Almost all cases of PWS can be confirmed by one of the above tests. However, in the rare event that laboratory tests do not confirm PWS, a clinical diagnosis can be helpful for the development of a management plan.

15
Treatment PWS cannot be cured. But, early intervention can help people build skills for adapting to the disorder. Early diagnosis can also help parents learn about the condition and prepare for future challenges. A health care provider can do a blood test to check for PWS. Exercise and physical activity can help control weight and help with motor skills. Speech therapy may be needed to help with oral skills. Human growth hormone has been found to be helpful in treating PWS. It can help to increase height, decrease body fat, and increase muscle mass. However, no medications have yet been found to control appetite in those with PWS.

16
In the Future To do more researches about Prader-Willi Syndrome and find out others genes related to it. To do a natural experiment of PWS in order to better understand the disease.

17
References The website of the Prader-Willi Syndrome Association(UK) http://pwsa.co.uk/main.php?catagory=1 http://pwsa.co.uk/main.php?catagory=1 http://www.pwsa.co.uk/main.php?catagory=1&sub_catagory=1 National Center for Biotechnology Information http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=gnd.section.182& ref=toc http://www.ncbi.nlm.nih.gov/books/bv.fcgi?rid=gnd.section.182& ref=toc http://www.ncbi.nlm.nih.gov/gene/4692?ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum http://www.ncbi.nlm.nih.gov/gene/4692?ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowS ection&rid=gnd.section.287 http://www.ncbi.nlm.nih.gov/books/bv.fcgi?call=bv.View..ShowS ection&rid=gnd.section.287 http://www.ncbi.nlm.nih.gov/sites/entrez?a=gene&Cmd=ShowD etailView&TermToSearch=7298 http://www.ncbi.nlm.nih.gov/sites/entrez?a=gene&Cmd=ShowD etailView&TermToSearch=7298 http://www.ncbi.nlm.nih.gov/gene/6638?ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum http://www.ncbi.nlm.nih.gov/gene/6638?ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
 catagory=1 catagory=1 catagory=1&sub_catagory=1 National Center for Biotechnology Information rid=gnd.section.182& ref=toc rid=gnd.section.182& ref=toc ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum call=bv.View..ShowS ection&rid=gnd.section call=bv.View..ShowS ection&rid=gnd.section.287 a=gene&Cmd=ShowD etailView&TermToSearch= a=gene&Cmd=ShowD etailView&TermToSearch=7298 ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum ordinalpos=1&itool=Ent rezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum")
18
Acknowledgements Ms. McMahon Prof. Brennan Dr. Sat Harlem Children Society National Library of Medicine Bronx Community College

Similar presentations














 imprinting Regulation of gene expression Developmental programming.>")







