Download presentation
Presentation is loading. Please wait.

1
Physics 250-06 “Advanced Electronic Structure” Density Functional Molecular Dynamics Contents: 1. Methods of Molecular Dynamics 2. Car-Parrinello Method. 3. Examples of Simulations. 4. Challenges and Future Developments For a review, see, Ab initio molecular dynamics: Theory and Implementation by Dominik Marx and Jurg Hutter Modern Methods and Algorithms of Quantum Chemistry, J. Grotendorst (Ed.), John von Neumann Institute for Computing, Julich, NIC Series, Vol. 1, pp. 301-449 (2000) (available on the WEB)
, John von Neumann Institute for Computing, Julich, NIC Series, Vol. 1, pp (2000) (available on the WEB).")
2
Foundations: Motions of Nuclei Non-relativistic hamiltonian including nuclei and electrons Wave functions become functionals of electronic and nuclear degrees of freedom Born-Oppenheimer approximation separates slow and fast degrees of freedom (M R >>m e )
")
3
Molecular Dynamics Classical equations of motions for the nuclei need to be solved where forces on nuclei are given by the derivative of the average total electronic energy with respect to the nucleir position

4
Forces Thus, integrating classical equation of motions we will be able to predict trajectories of atoms and answer the questions of equilibrium atomic configurations, melting temperatures, phase diagrams, diffusions processes, and so on. The major difficulty is to find forces which evolve as nuclear coordinates change their positions:

5
Classical Molecular Dynamics In classical molecular dynamics, the average electronic energy as a functional of nuclear coordinates is parametrized in terms of various (one-,two-,three-,etc- body) interatomic potentials which, for example, can be found using various fits to observable properties.
 interatomic potentials which, for example, can be found using various fits to observable properties.")
6
Typical two-body interactions (Lennard-Jones van der Waals), look like that Equilibrium
, look like that Equilibrium")
7
Simulation of interaction energy of argon dimer is represented by van der Waals interactions

8
Dimensionality bottleneck Decomposition of global potential energy onto pair-like potentials is crucial if one wants to avoid dimensionality bottleneck. 500 argon atoms need a fit of a function of 1500 coordinates while pair-potential representation needs a fit of a function in one dimension only:

9
Ab Initio Molecular Dynamics In ab initio molecular dynamics, the average electronic energy as a functional of nuclear coordinates is recomputed using quantum mechanical methods such as Density Functional Theory There are two intrinsically different methods to integrate electronic degrees of freedom while performing simulations: Born-Oppenheimer Molecular Dynamics Ehrenfest Molecular Dynamics

10
Born-Oppenheimer Molecular Dynamics In Born-Oppenheimer molecular dynamics, electronic structure problem is solved self-consistently each time for a given set of configurations of nuclei In other words electrons are allowed to fully relax and reach its minimum for a given position of nuclei.

11
Consider calculation of force using Hellmann-Feynman Theorem

12
Recall the expression for the hamiltonian its derivative with respect to R is trivial and the force is given by where we used a defintion of electronic density:

13
Remarkably that the electronic contribution to the force on a given nuclei is represented by a classical electrostatic force made by electronic charge density cloud on the charge Z R centered at R : Thus, density functional theory is ideally suited for performing ab initio molecular dynamics simulations!

14
Born-Oppenheimer molecular dynamics simulations using DFT involve the following steps: 1. Fix positions of nuclei, {R 1 …R N }, solve DFT equations self-consistently 2. Find electrostatic force on each atom: 3. Integrate equation of motion for the nuclei: perform a time step and find new positions of nuclei MD Simulation

15
Born-Oppenheimer molecular dynamics became popular since many electronic structure codes are available Obvious drawback: necessity to fully relax electronic subsystem while moving the atoms. This makes it computationally very slow. Full self-consistency at each MD step may not be necessary especially when system is far from its equilibrium, one simply needs a rough idea on the force field for a given atomic configuration Construction of potential energy surface is avoided since forces are found “on the fly”

16
Ehrenfest Molecular Dynamics In Ehrenfest molecular dynamics, electronic structure problem is solved using time-dependent Schrodinger equation and both equations for nuclei and electrons are solved simultaneously Construction of potential energy surface is avoided by finding forces “on the fly”

17
Let expand the electronic wave functions into full set of Slater determinants Thus, Ehrenefest molecular dynamics tracks the time dependence of the coefficients c k (t) as the system evolves with time. The corresponding equations of motions read as Equations are coupled via Thus the approach includes all non-adiabatic transitions between various states E k during nuclear motions

18
If only one state with the lowest energy is kept, we obtain which needs to be integrated simultaneously since the hamiltonian depends on time via nuclear coordinates which are time dependent! The last point makes the Ehrenfest molecular dynamics fundamentally different from Born-Oppenheimer molecular dynamics.

19
Ehrenfest molecular dynamics simulations using time dependent version of DFT involve simultaneous integration: where electrostatic force on each atom can be evaluated:

20
Comparing Ehrenfest molecular dynamics simulations using time dependent DFT and Born-Oppenheimer molecular dynamics using static DFT we see that in TD-DFT Kohn-Sham states are represented by frequency integrals while in static DFT frequency-dependent functions are peaked at a given Kohn-Sham eigenvalue:

21
The main task achieved in Ehrenfest dynamics is simply to keep the wavefunction automatically minimized as the nuclei are propagated. This, however, might be achieved in principle by another sort of dynamics, namely Car-Parrinello molecular dynamics. In summary, the “Best of all Worlds Method" should (i) integrate the equations of motion on the (long) time scale set by the nuclear motion but nevertheless (ii) take intrinsically advantage of the smooth time evolution of the dynamically evolving electronic subsystem as much as possible. The second point allows to circumvent explicit diagonalization or minimization to solve the electronic structure problem for the next molecular dynamics step as it is done in Born-Oppenheimer Molecular dynamics. Car-Parrinello molecular dynamics is an efficient method to satisfy requirement (ii) in a numerically stable fashion and makes an acceptable compromise concerning the length of the time step (i). Car-Parrinello Molecular Dynamics
 integrate the equations of motion on the (long) time scale set by the nuclear motion but nevertheless (ii) take intrinsically advantage of the smooth time evolution of the dynamically evolving electronic subsystem as much as possible. The second point allows to circumvent explicit diagonalization or minimization to solve the electronic structure problem for the next molecular dynamics step as it is done in Born-Oppenheimer Molecular dynamics. Car-Parrinello molecular dynamics is an efficient method to satisfy requirement (ii) in a numerically stable fashion and makes an acceptable compromise concerning the length of the time step (i). Car-Parrinello Molecular Dynamics.")
22
In CPMD a two-component quantum / classical problem is mapped onto a two-component purely classical problem with two separate energy scales at the expense of loosing the explicit time dependence of the quantum subsystem dynamics. Now, in classical mechanics the force on the nuclei is obtained from the derivative of a Lagrangian with respect to the nuclear positions. This suggests that a functional derivative with respect to the orbitals, which are interpreted as classical fields, might yield the force on the orbitals, given a suitable Lagrangian. In addition, possible constraints within the set of orbitals have to be imposed, such as e.g. orthonormality (or generalized orthonormality conditions that include an overlap matrix).
..")
23
Car-Parrinello Lagrangian Car and Parrinello (1985) have postulated the following Lagrangian where i are fictitious masses and i are classical fields. Classical action needs to be minimized which results in equation of motions Kinetic EnergyPotential EnergyConstraints

24
Car-Parrinello Equations of Motions Car and Parrinello (1985) have derived equations of motions which are obviously transformed back to Born-Oppenheimer molecular dynamics if fictitious masses for the electrons i At the eqilibrium, there are no forces on electrons, therefore Ground state of density functional theory is reached with the eigenvalues being the Kohn Sham eigenstates.
 have derived equations of motions which are obviously transformed back to Born-Oppenheimer molecular dynamics if fictitious masses for the electrons i At the eqilibrium, there are no forces on electrons, therefore Ground state of density functional theory is reached with the eigenvalues being the Kohn Sham eigenstates.")
25
Why does the Car-Parrinello Method works? Conserved Energy in CPMD is not a physical energy but supplemented with a small fictitious kinetic term

26
Various Energies extracted from CPMD for a model system. T fict E DFT The fictitious kinetic energy of the electrons is found to perform bound oscillations around a constant, i.e. the electrons do not heat up“ systematically in the presence of the nuclei; note that T fict is a measure for deviations from the exact Born-Oppenheimer surface. Closer inspection shows actually two time scales of oscillations: the one visible in the Figure stems from the drag exerted by the moving nuclei on the electrons and is the mirror image of the E DFT fluctuations. As a result the physical energy (the sum of the nuclear kinetic energy and the electronic total energy which serves as the potential energy for the nuclei) is essentially constant on the relevant energy and time scales.
 is essentially constant on the relevant energy and time scales..")
27
Given the adiabatic separation and the stability of the propagation, the central question remains if the forces acting on the nuclei are actually the “correct" ones in Car-Parrinello molecular dynamics. As a reference serve the forces obtained from full self-consistent minimizations of the electronic energy at each time step, i.e. Born- Oppenheimer molecular dynamics with extremely well converged wavefunctions.

28
How to control adiabaticity? Since the electronic degrees of freedom are described by much heavier masses than the electronic masses, time step to perform CPMD simulations needs not to be too small as compared to Ehrenfest molecular dynamics. For a system with a gap in the spectrum, the lowest possible frequency of “fictitious” electronic oscillations To guarantee adiabatic separation this frequency should be much larger than the typical phonon energy and/or the gap in the spectrum which would make sure that the electrons follow the nuclei adiabatically. Hence fictitious mass .

29
At the same time, small fictitious mass would imply smaller and smaller time step because maximum fictitious electronic frequency is proportional to the plane-wave cutoff energy As a result a compromise fictitious mass needs to be found in CPMD simulations. For metals gap is zero and zero frequency “fictitious” electronic modes occur in the spectrum overlapping with the phonon spectrum. Thus, a well-controlled Born-Oppenheimer approach can only be recommended

30
Consider variational principle which can be used to find an upper bound for the lowest eigenstates of the hamiltonian using basis set expansion CP Method as dynamical solution of DFT equations CP Method invented a new way to solve Kohn-Sham equations alternative to diagonalization. CPMD offers a way to determine the coefficients without reduction to the eigenvalue problem.

31
In traditional molecular dynamics the system heated at temperature T is gradually cooled and find its minimum energy configuration. (after Payne et.al, RMP 1992)
.")
32
In CMPD scheme the total energy is a functional of the coefficients which expands the wave function in some basis set. Each coefficient is regarded as a coordinate of a classical particle. To minimize the KS functional each particle is given some kinetic energy and the system is gradually cooled Until the set of “coordinates” c reaches its values that minimize the functional. Thus the problem of solving KS equations is reduced to solving “fictitious” classical equations of motions

33
(after Payne et.al, RMP 1992)
")
34
Flowchart of the CPMD algorithm (Payne et.al, RMP 1992)
")
35
How KS states converge: (CP, PRL 1985)
")
36
What about Hellmann-Feynman forces? Derivation assumes that the wave functions are exact solutions of the Schrodinger equation. On the language of the DFT The last contribution is equal to zero only if self-consistency is reached

37
Non-Self-Consistency Force In general, the force is made of Hellmann-Feynman contribution and contribution due to non-self-consistency (also due to incompleteness of the basis set which we discuss later): So, in Born-Oppenheimer MD, well convergent self-consistent calculations are needed to eliminate the non-self-consistency correction.
: So, in Born-Oppenheimer MD, well convergent self-consistent calculations are needed to eliminate the non-self-consistency correction.")
38
The crucial point is, however, that in Car-Parrinello as well as in Ehrenfest molecular dynamics it is not the minimized expectation value of the electronic Hamiltonian that yields the consistent forces. What is merely needed is to evaluate the expression with the Hamiltonian and the associated wavefunction available at a certain time step In other words, it is not required (concerning the present discussion of the contributions to the force!) that the expectation value of the electronic Hamiltonian is actually completely minimized for the nuclear configuration at that time step. Whence, full self-consistency is not required for this purpose in the case of Car-Parrinello (and Ehrenfest) molecular dynamics. As a consequence, the non-self-consistency correction to the force is irrelevant in Car-Parrinello (and Ehrenfest) simulations. Heuristically one could also argue that within Car-Parrinello dynamics the non- vanishing non-self-consistency force is kept under control or counterbalanced by the non-vanishing “mass times acceleration term" which is small but not identical to zero and oscillatory. This is sufficient to keep the propagation stable, whereas, i.e. an extremely tight minimization, is required by its very definition in order to make the Born-Oppenheimer approach stable. Thus, also from this perspective it becomes clear that the fictitious kinetic energy of the electrons is a measure for the departure from the exact Born-Oppenheimer surface during Car-Parrinello dynamics.
 that the expectation value of the electronic Hamiltonian is actually completely minimized for the nuclear configuration at that time step. Whence, full self-consistency is not required for this purpose in the case of Car-Parrinello (and Ehrenfest) molecular dynamics. As a consequence, the non-self-consistency correction to the force is irrelevant in Car-Parrinello (and Ehrenfest) simulations. Heuristically one could also argue that within Car-Parrinello dynamics the non- vanishing non-self-consistency force is kept under control or counterbalanced by the non-vanishing mass times acceleration term which is small but not identical to zero and oscillatory. This is sufficient to keep the propagation stable, whereas, i.e. an extremely tight minimization, is required by its very definition in order to make the Born-Oppenheimer approach stable. Thus, also from this perspective it becomes clear that the fictitious kinetic energy of the electrons is a measure for the departure from the exact Born-Oppenheimer surface during Car-Parrinello dynamics..")
39
Error Cancellation in Hellmann-Feynman forces

40
Publications on Molecular Dynamics

41
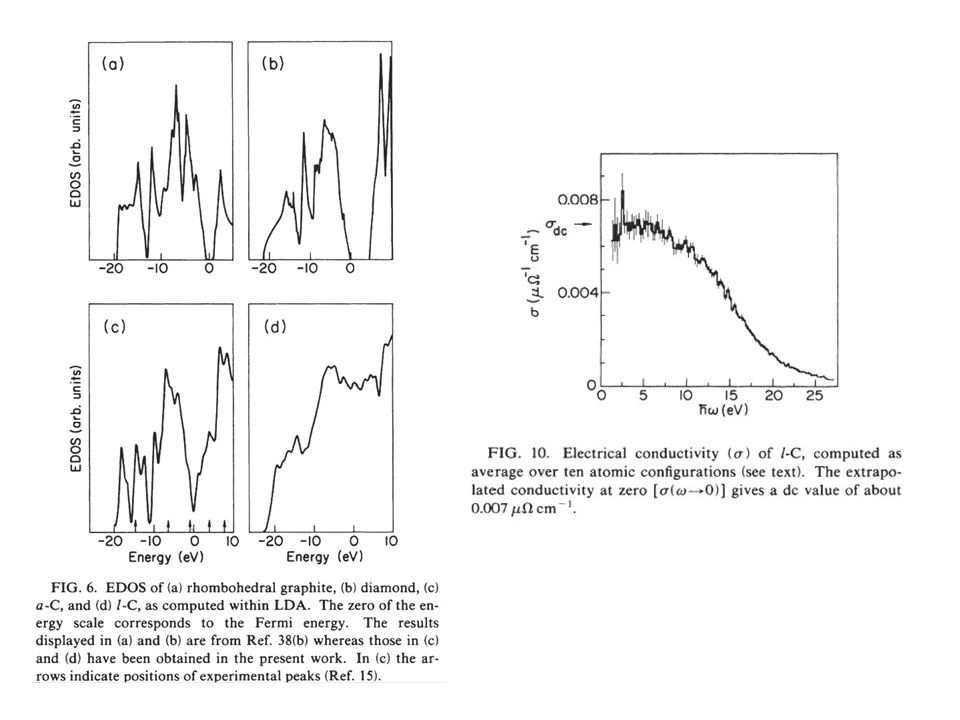
Examples of Simulations: Carbon (Galli, et.al)
")
43
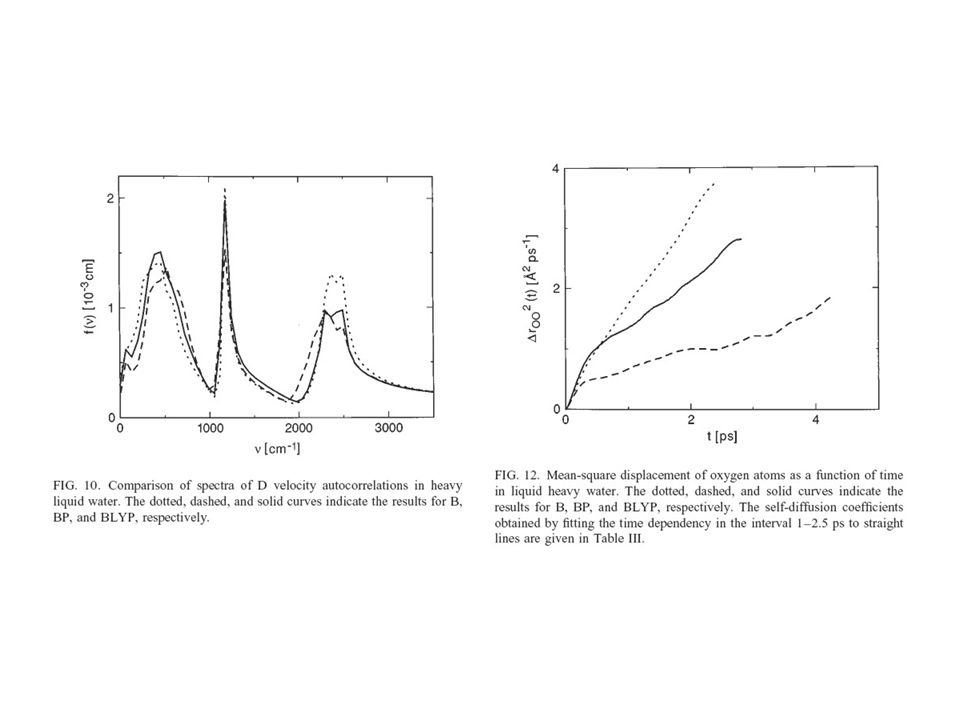
Examples of Simulations: Water (Sprik et.al.)
")
45
Current Challenges Simulations with basis sets different from plane waves. Difficulties in determining forces. Molecular dynamics of strongly correlated systems: better than DFT functionals needed. Again, d-, and f-electrons are better represented with local orbital basis sets, determinations of forces would complicate the simulations.

Similar presentations



 Workshop.>")
















