Download presentation
Presentation is loading. Please wait.

1
Comparative Protein Structure Modeling Lecture 4.1
Roberto Sanchez Structural Biology Program, Mount Sinai School of Medicine New York, NY 10029, USA Overview of the talk. I will explain what CM is and how and why it works. I’ll show examples of single protein (manual) modeling which make certain point with respect to the advantages, limitations and applications of CM. Automated large-scale modeling will be described in the context of structural genomics. The ModBase database will be briefly described. What is comparative modeling and why is it useful? Steps in CM (overview + some details) Accuracy of comparative models Loop modeling CM and Structural Genomics
 modeling which make certain point with respect to the advantages, limitations and applications of CM. Automated large-scale modeling will be described in the context of structural genomics. The ModBase database will be briefly described. What is comparative modeling and why is it useful Steps in CM (overview + some details) Accuracy of comparative models. Loop modeling. CM and Structural Genomics.")
2
Function via Structure
GFCHIKAYTRLIM… Sequence Structure Function Physically, function is determined by the protein’s structure and its dynamics. Thus, we are interested in characterizing function of a protein sequence based on its three-dimensional structure. But there are several problems: First, we do not know 3D structure of most proteins, only 10,000 proteins have had their structure determined, while about 500,000 protein sequences are known. Second, even knowing the structure of a protein is frequently not sufficient to predict its functional properties. Nevertheless, structural biology has demonstrated that knowing structure of a protein is a very good thing. So, how do we get all these structures? Not by experiment. Thus, by prediction.

3
Why is it useful to know the structure of a protein not only its sequence?
The biochemical function (activity) of a protein is defined by its interaction with other molecules. The biological function is in large part a consequence of these interactions. The 3D structure is more informative than sequence because interactions are determined by residues that are close in space but are frequently distant in sequence. In addition, since evolution tends to conserve function and function depends more directly on structure than on sequence, structure is more conserved in evolution than sequence. The net result is that patterns in space are frequently more recognizable than patterns in sequence.
 of a protein is defined by its interaction with other molecules. The biological function is in large part a consequence of these interactions. The 3D structure is more informative than sequence because interactions are determined by residues that are close in space but are frequently distant in sequence. In addition, since evolution tends to conserve function and function depends more directly on structure than on sequence, structure is more conserved in evolution than sequence. The net result is that patterns in space are frequently more recognizable than patterns in sequence.")
4
Why Protein Structure Prediction?
Known Sequences (5/30/01) : 694,000 Known Structures (5/29/01) : ,200 Comparative Modeling is a Protein Structure Prediction Method We know the experimental 3D structure for less than 3% of the protein sequences. For the remaining 97% we need some sort of 3D structure prediction.
 : 694,000. Known Structures (5/29/01) : 15,200. Comparative Modeling is a Protein Structure Prediction Method. We know the experimental 3D structure for less than 3% of the protein sequences. For the remaining 97% we need some sort of 3D structure prediction.")
5
What is Comparative Protein Structure Modeling?
Protein Structure Prediction …SDVIFTEDGILICNRK… Comparative Modeling is a Protein Structure Prediction Method

6
Principles of Protein Structure
GFCHIKAYTRLIMVG… Folding Anabaena 7120 Anacystis nidulans Condrus crispus Desulfovibrio vulgaris Evolution There are two sets of principles that proteins follow. Physical and evolutionary. Examples. Challenge is to unify them. We try that. Ab initio prediction Fold Recognition Comparative Modeling

7
Steps in Comparative Protein Structure Modeling
Template Search TEMPLATE START ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPERASFQWMNDK TARGET No Target – Template Alignment MSVIPKRLYGNCEQTSEEAIRIEDSPIV---TADLVCLKIDEIPERLVGE ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPE Model Building Main steps in comparative modeling, all approaches: 1) Threading can be used, others, in step 1. Model Evaluation OK? END Yes A. Šali, Curr. Opin. Biotech. 6, 437, 1995. R. Sánchez & A. Šali, Curr. Opin. Str. Biol. 7, 206, 1997. M. Marti et al. Ann. Rev. Biophys. Biomolec. Struct., 29, 291, 2000.
 Threading can be used, others, in step 1. Model Evaluation. OK END. Yes. A. Šali, Curr. Opin. Biotech. 6, 437, R. Sánchez & A. Šali, Curr. Opin. Str. Biol. 7, 206, M. Marti et al. Ann. Rev. Biophys. Biomolec. Struct., 29, 291,")
8
Template Search Methods
Sequence similarity searches (BLAST, FastA) Profile and iterative methods (HMMs, PSI-BLAST) Structure based threading (THREADER, PROFIT)
 Profile and iterative methods (HMMs, PSI-BLAST) Structure based threading (THREADER, PROFIT)")
9
Target – Template Alignment Methods
Dynamic Programming Pairwise Alignments Multiple Alignments, Profiles, HMMs Structure based approaches (Threading)
")
10
Model Building Methods
Rigid Body Assembly (COMPOSER) Segment Matching (SEGMOD) Satisfaction of Spatial Restraints (MODELLER) A. Šali, Curr. Opin. Biotech. 6, 437, 1995 R. Sánchez & A. Šali, Curr. Opin. Str. Biol. 7, 206, 1997
 Segment Matching (SEGMOD) Satisfaction of Spatial Restraints (MODELLER) A. Šali, Curr. Opin. Biotech. 6, 437, R. Sánchez & A. Šali, Curr. Opin. Str. Biol. 7, 206,")
11
Comparative Modeling by MODELLER
3D GKITFYERGFQGHCYESDC-NLQP SEQ GKITFYERG---RCYESDCPNLQP EXTRACT Spatial Restraints F(R) = Ppi(fi/I) i SATISFY Spatial Restraints A. Šali & T. Blundell, J. Mol. Biol. 234, 779, 1993
 = Ppi(fi/I) i. SATISFY. Spatial Restraints. A. Šali & T. Blundell, J. Mol. Biol. 234, 779,")
12
Model Evaluation methods
Stereochemistry (PROCHECK) Environment (Profiles3D) Statistical potentials based methods (PROSAII)
 Environment (Profiles3D) Statistical potentials based methods (PROSAII)")
13
Model Evaluation: Alignment Errors
R. Sánchez & A. Šali, Proteins, Suppl. 1, 50-58, 1997

14
Are models useful if they are just copies of the template?

15
Do mast cell proteases bind proteoglycans? Where? When?
Predicting features of a model that are not present in the template mMCPs bind negatively charged proteoglycans through electrostatic interactions? Comparative models used to find clusters of positively charged surface residues. Tested by site-directed mutagenesis.. Huang et al. J. Clin. Immunol. 18,169,1998. Matsumoto et al. J.Biol.Chem. 270,19524,1995. Šali et al. J. Biol. Chem. 268, 9023, 1993. GRASP (Honig). Some members have His, some do not. Simple criteria resulting in concrete predictions. Models based on about 35% sequence identity to trypsin. But trypsin, the template, does not bind proteglycans. Native mMCP-7 at pH=5 (His+) Native mMCP-7 at pH=7 (His0)
. Some members have His, some do not. Simple criteria resulting in concrete predictions. Models based on about 35% sequence identity to trypsin. But trypsin, the template, does not bind proteglycans. Native mMCP-7 at pH=5 (His+) Native mMCP-7 at pH=7 (His0)")
16
Model Accuracy

17
Typical Errors in Comparative Models
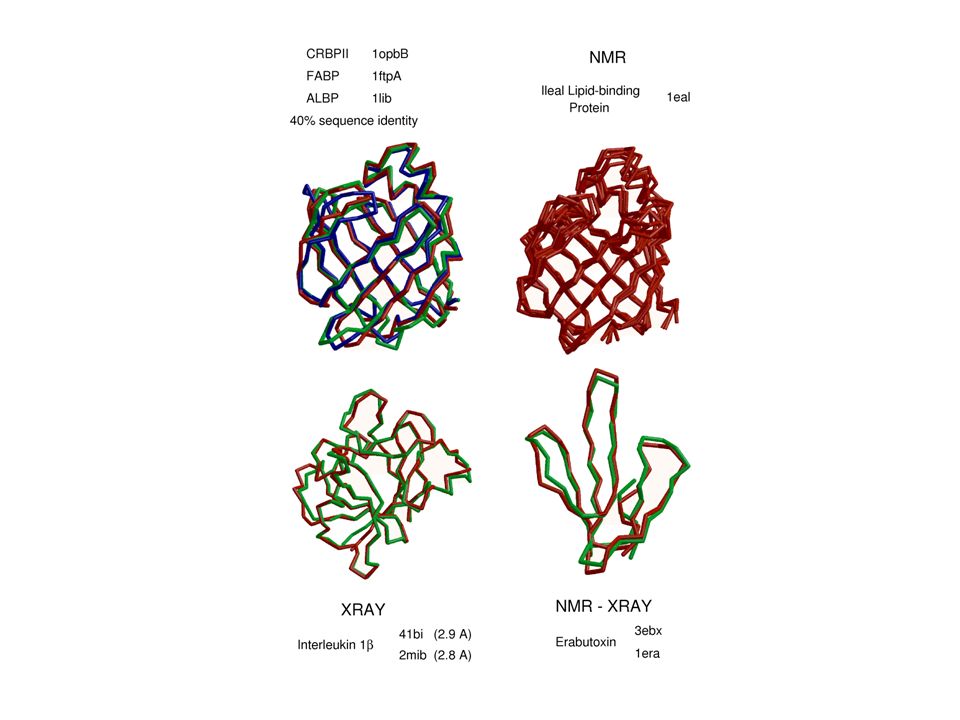
Incorrect template MODEL X-RAY TEMPLATE Misalignment Application of comparative modeling to proteins of known structure identifies the following five types of errors in comparative models. Template selection (<25% sequence identity). Misalignments (<35% sequence identity). Loop modeling, shifts, sidechain modeling (whole range). Region without a template Distortion in correctly aligned regions Sidechain packing
. Misalignments (<35% sequence identity). Loop modeling, shifts, sidechain modeling (whole range). Region without a. template. Distortion in correctly. aligned regions. Sidechain packing.")
18
CASP: Lessons from Blind Predictions
Build models for proteins of unknown structure. Structures are determined after the models are submitted. Models are evaluated by comparing them with the corresponding experimental structures.

19
CASP: Lessons from Blind Predictions Multiple Template Models
Comparative modeling (by MODELLER) can combine the best regions from each template. The per-residue accuracy of comparative models can not be higher than that of any of the templates. The overall accuracy of models can be higher than that of any of the templates.
 can combine the best regions from each template. The per-residue accuracy of comparative models can not be higher than that of any of the templates. The overall accuracy of models can be higher than that of any of the templates.")
20
CASP: Lessons from Blind Predictions (DFR)
R. Sánchez & A. Šali, Proteins, Suppl. 1, 50-58, 1997

21
Model Accuracy as a Function of Target-Template Sequence Identity
The individual errors integrate into overall errors. It is good to be able to assess the overall accuracy of a model. It is not so bad if there are errors in a model, as long as one knows that and takes them into account when the model is used. A useful indicator of the overall structural error is a measure of sequence similarity between the modeled protein sequence and the sequence of the known template structure. The reason is … A simple sequence similarity measure is sequence identity. This plot shows … It was obtained by calculating automatically approx 10,000 models for proteins of known structure and by comparing these models with the actual structures. Describe the lower curve. Describe the upper curve. Mention the criticism of CM that it does not improve the template – this is what it refers to. But it is not a fair criticism because we do not know the correct target-template alignment in the absence of the target structure, and in any case even a model with a worse RMSD than the template can be more useful than the template to learn about the function of a protein, as I will demonstrate later.

23
Some Models Can Be Surprisingly Accurate (in Some Regions)
24% sequence identity YJL001W 1rypH 25% sequence identity YGL203C 1ac5 Ser 176 His 488 Asp 383 The fact that we have some models and that a fraction is very accurate, if we can detect those, we can do surprisingly great things. Two examples of models calculated before the structure was known.

25
Applications of Comparative Models
It is convenient to divide comparative models into three classes, based on their predicted overall accuracy. Applications depend on accuracy. Applications may or may not succeed. Accuracy of a model needs to be predicted and considered before the model is used. Even low resolution models can be used for some questions. And even the highest resolution models, even x-ray structures, are not accurate enough for some questions (eg, catalysis).
.")
26
Loop Modeling in Protein Structures
a+b barrel: flavodoxin antiparallel b-barrel IG fold: immunoglobulin I will now describe in more detail a significant methodological improvement in only one area: Loop modeling. Loops are important for function. The size of a problem – loop length. We will be modeling individual loops here, without the presence of a ligand (induced fit), and without being particularly concerned with the dynamics of loops, though I will make some comments about it. A. Fiser, R. Do & A. Šali, Prot. Sci., 9, 1753, 2000
, and without being particularly concerned with the dynamics of loops, though I will make some comments about it. A. Fiser, R. Do & A. Šali, Prot. Sci., 9, 1753,")
27
Loop modeling strategies
Database search Conformational search database is complete only up to 4-6 residues even in DB search, the different conformations must be ranked loops longer than 4 residues need extensive optimization DB method is efficient for specific families (eg. Canonical loops in Ig’s, b- hairpins etc)
")
28
Loop Modeling by Conformational Search
Conformational search more general than database searches. Three components: Standard protein representation. Scoring function is the key. Optimization by MD/SA: bake and shake, many times. Protein representation. Energy (scoring) function. Optimization algorithm.
 function. Optimization algorithm.")
29
Energy Function for Loop Modeling
The energy function is a sum of many terms: 1) Statistical preferences for dihedral angles: 2) Restraints from the CHARMM-22 force field: Flexibility of MODELLER! 3) Statistical potential for non-bonded contacts:
 Statistical preferences for dihedral angles: 2) Restraints from the CHARMM-22 force field: Flexibility of MODELLER! 3) Statistical potential for non-bonded contacts:")
30
Mainchain Terms for Loop Modeling
In combination with the non-bonded terms, this is the key term that allowed us to improve the accuracy of loop models significantly.

31
Optimization of Objective Function

32
Calculating an Ensemble of Loop Models
Stochastic optimization, thus the need for many independent optimizations. One discovers two basic situations: similar solutions, dissimilar solutions. But which one prevails? One needs a lot of different loops to test the method, calculate averages.

33
Accuracy of loop models
Accuracy versus number of optimisation, length of loop and range of distortion

34
Assessing Accuracy of Loop Models
As for the whole models, it is important to predict the error of the loop model. This can be achieved accurately by comparing the structural similarity of several lowest energy solutions, obtained from the independent modeling predictions of the same loop.

35
Accuracy of Loop Modeling
RMSD=0.6Å HIGH ACCURACY (<1Å) 50% (30%) of 8-residue loops RMSD=1.1Å MEDIUM ACCURACY (<2Å) 40% (48%) of 8-residue loops RMSD=2.8Å LOW ACCURACY (>2Å) 10% (22%) of 8-residue loops Out of rigorous statistical evaluation, measuring the accuracy of the method as a function of a variety of variables. Environment accuracy! This results reduce the average RMSD error to about one half of that of what I think was previously the most accurate method. A. Fiser, R. Do & A. Šali, Prot. Sci., 9, 1537, 2000
 50% (30%) of 8-residue loops. RMSD=1.1Å. MEDIUM ACCURACY (<2Å) 40% (48%) of 8-residue loops. RMSD=2.8Å. LOW ACCURACY (>2Å) 10% (22%) of 8-residue loops. Out of rigorous statistical evaluation, measuring the accuracy of the method as a function of a variety of variables. Environment accuracy! This results reduce the average RMSD error to about one half of that of what I think was previously the most accurate method. A. Fiser, R. Do & A. Šali, Prot. Sci., 9, 1537,")
36
Fraction of Loops Modeled With at Least Medium Accuracy
For up to 8 residue loops, when the environment is approximately correct, the loop modeling problem in the narrow sense is now essentially solved. However, in practice there are complications: environment is not always correctly modeled decreases loop accuracy; in fact, sometimes we need to model several neighboring loops at the same time. It is difficult to decide which segments to model ab initio because they are different from the template. And as I said before, we are modeling average conformations – no dynamics, which is frequently important for function. Also, no ligands here, so no induced fit is modeled. Nevertheless, this should prove useful for low resolution single ligand docking methods or for computational methods that produce high-quality ligand libraries.

37
Problems in Practical Loop Modeling
Decide which regions to model as loops. Correct alignment of anchor regions & environment. Modeling of a loop. In practice, there are some additional problems in loop modeling … T0076: 46-53 RMSDmnch loop = 1.37 Å RMSDmnch anchors = 1.52 Å T0058: 80-85 RMSDmnch loop = 1.09 Å RMSDmnch anchors = 0.29 Å

38
How can Comparative Modeling be used in Structural Genomics?

39
Structural Genomics Definition: The aim of structural genomics is to put every protein sequence within a modeling distance of a known protein structure. Size of the problem: There are a few thousand domain fold families. There are ~20,000 sequence families (30% sequence id). Solution: Determine many protein structures. Increase modeling distance. Moving to genomes and large numbers of models now, from individual What is SG. Base projection on the current numbers. Collaborators. NYSGRC. Šali. Nat. Struct. Biol. 5, 1029, Burley et al. Nat. Genet. 23, 151, 1999. Šali & Kuriyan. TIBS 22, M20, Sanchez et al. Nat. Str. Biol. 7, 986, 2000
. Solution: Determine many protein structures. Increase modeling distance. Moving to genomes and large numbers of models now, from individual What is SG. Base projection on the current numbers. Collaborators. NYSGRC. Šali. Nat. Struct. Biol. 5, 1029, Burley et al. Nat. Genet. 23, 151, Šali & Kuriyan. TIBS 22, M20, Sanchez et al. Nat. Str. Biol. 7, 986,")
40
How can Comparative Modeling be used in Structural Genomics?
Target Selection How many structures need to be solved? Which structures should we solve first? Target Amplification How much of the sequence space is covered by: a new structure all structures

41
Target Selection for Structural Genomics
Select targets such that every protein sequence is within a modeling distance of a known protein structure. Modeling distance: correct alignment, corresponding to >30% sequence identity. G. Kurban, R. Sánchez, A. Šali, T. Gaasterland.

42
Models + Fold Assignments
Leveraging Templates by Comparative Modeling Quantifying Productivity of Structural Genomics Modeling Template Models + Fold Assignments Reliable Models Accurate Models Less Accurate Models Fold Assignments P007 27 19 9 P008 18 11 7 P018 108 32 3 29 76 P100 26 12 5 14 Total 179 89 38 51 90 Models are in MODBASE at

43
MODPIPE: Large-Scale Comparative Protein Structure Modeling
START For each sequence END 1 For each template Prepare PSI-BLAST PSSM by comparing the sequence against the NR database of sequences Align the matched part of the target sequence with the template structure PSI-BLAST MODELLER Use the sequence PSSM to search against the representative set of PDB chains (F and no-F) Build a model for the target segment by satisfaction of spatial restraints Comparative Modeling is a Protein Structure Prediction Method Use the PDB chain PSSMs to search against the sequence (F and no-F) Evaluate the model Select Templates using a permissive E-value cutoff R. Sánchez & A. Šali, Proc. Natl. Acad. Sci. USA 95, 13597, 1998 R. Sánchez, F. Melo, N. Mirkovic, A. Šali, in preparation
 Build a model for the target segment by satisfaction of spatial restraints. Comparative Modeling is a Protein Structure Prediction Method. Use the PDB chain PSSMs to search against the sequence (F and no-F) Evaluate the model. Select Templates using a permissive E-value cutoff. R. Sánchez & A. Šali, Proc. Natl. Acad. Sci. USA 95, 13597, R. Sánchez, F. Melo, N. Mirkovic, A. Šali, in preparation.")
44
MODPIPE Model of Yeast Hypothetical Protein YIL073C
PDB 1a17 template YIL073C model We searched for very good models from an energetic point of view, based on low sequence similarity to the template – non-trivial matches, that is surprises. This one makes good biological sense, illustrates what one can sometimes do. About 5% of all matches are in this class (500 in the case of the yeast genome). Fold assignment: from a random sequence of 20 characters to quite a specific prediction of function and in general also experiments to test the hypothesis. This illustrates a major new problem for bioinformatics – informing the relevant people about the predictions. E-value = 65 Seq. Id. = 20% pG = 0.97 Das et al. EMBO J. 17, 1192, 1998 The tetratricopeptide repeat (TPR) is a degenerate 34 aa sequence identified in a variety of proteins, present in tandem arrays, mediates protein-protein interactions. R. Sánchez, F. Melo, N. Mirkovic, A. Šali.
. Fold assignment: from a random sequence of 20 characters to quite a specific prediction of function and in general also experiments to test the hypothesis. This illustrates a major new problem for bioinformatics – informing the relevant people about the predictions. E-value = 65. Seq. Id. = 20% pG = Das et al. EMBO J. 17, 1192, The tetratricopeptide repeat (TPR) is a degenerate 34 aa sequence identified in a variety of proteins, present in tandem arrays, mediates protein-protein interactions. R. Sánchez, F. Melo, N. Mirkovic, A. Šali.")
45
Mycoplasma genitalium MODPIPE Models
Number of ORFs 479 Average ORF length 364 Number of ORFs modeled 477 (99%) ORFs with fold assignment (PSI-BLAST hit or model) 330 (69%) ORFs with reliable models 273 (57%) not based on PSI-BLAST hit 76 ( 16%) Average model size 176 Average sequence identity 28.7%
 ORFs with fold assignment. (PSI-BLAST hit or model) 330 (69%) ORFs with reliable models. 273 (57%) not based on PSI-BLAST hit. 76 ( 16%) Average model size Average sequence identity. 28.7%")
46
Mycoplasma genitalium MODPIPE Models
Number of ORFs 479 Average ORF length 364 Number of ORFs modeled 477 (99%) ORFs with fold assignment (PSI-BLAST hit or model) 330 (69%) ORFs with reliable models 273 (57%) not based on PSI-BLAST hit 76 ( 16%) Average model size 176 Average sequence identity 28.7%
 ORFs with fold assignment. (PSI-BLAST hit or model) 330 (69%) ORFs with reliable models. 273 (57%) not based on PSI-BLAST hit. 76 ( 16%) Average model size Average sequence identity. 28.7%")
47
Factors affecting coverage: PDB growth
New problem: comparative modeling has to be done entirely automatically because (i) used by non-experts; (ii) used efficiently by experts; (iii) used on a large-scale as a result of genome sequencing and structural genomics. Most of the protein for which some structural information will be available will be models, not actual structures. It is highly non-trivial to create a fully automated system comparable in reliability to human expert. Fold assignments Reliable models
 used by non-experts; (ii) used efficiently by experts; (iii) used on a large-scale as a result of genome sequencing and structural genomics. Most of the protein for which some structural information will be available will be models, not actual structures. It is highly non-trivial to create a fully automated system comparable in reliability to human expert. Fold assignments. Reliable models.")
48
Top 10 organism by number of models
Organism Statistics Top 10 organism by number of models Organism # sequences # models models/ seq# # CATH folds Homo sapiens 13,785 37,638 2.73 315 HIV type 1 25,654 33,180 1.29 12 D. melanogaster 8,248 25,314 3.06 299 C. elegans 7,260 20,095 2.76 289 A. thaliana 8,852 18,695 2.11 294 Mus musculus 6,232 17,248 271 R. norvegicus 3,586 9,299 2.59 246 S. cerevisiae 2,580 5,749 2.22 237 S. Pombe 2,315 4,497 1.94 221 E. coli 2,862 4,333 1.51 259

49
Top 10 organism by number of models
Organism Statistics Top 10 organism by number of models Organism Avg. seq. length Avg. model length Avg. Sequence coverage “Organism” coverage Homo sapiens 517 191 0.55 0.36 HIV type 1 165 124 0.84 0.75 D. melanogaster 634 209 0.47 0.32 C. elegans 563 0.50 0.37 A. thaliana 480 218 0.45 Mus musculus 510 0.53 R. norvegicus 511 207 0.57 0.40 S. cerevisiae 590 255 0.43 S. Pombe 527 247 0.58 0.46 E. coli 367 248 0.67

50
MODBASE R. Sánchez, U. Pieper, N.Mirkovic, P. I. W. de Bakker, E. Wittenstein, and A. Šali. Nucl. Acids Res., 28, R. Sánchez and A. Šali. Bioinformatics, 15, 1060, 1999

51
Review Comparative models can help in understanding the function of proteins by: Detecting remote structural (functional?) relationships. Revealing features that are not present in the templates. Revealing features that are not recognizable from the sequence. Insertions (loops) up to 8 residues long can be reliable modeled. Comparative modeling can play a role in structural genomics: in target selection and in amplifying the experimental data. At present, useful 3D models can be obtained for domains in approximately 50% of the proteins (25% of domains), because we improved our techniques and because of the many known protein structures and sequences. Graduate student thesis time scale and life will change.
 up to 8 residues long can be reliable modeled. Comparative modeling can play a role in structural genomics: in target selection and in amplifying the experimental data. At present, useful 3D models can be obtained for domains in approximately 50% of the proteins (25% of domains), because we improved our techniques and because of the many known protein structures and sequences. Graduate student thesis time scale and life will change.")
Similar presentations


 and connectivity.>")







 287, 797-815 2.Protein Fold Recognition by Prediction-based Threading Rost, B., Schneider,>")
.>")



>")
![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")



