Download presentation
Presentation is loading. Please wait.

1
What is Pharmacology? derived from the Greek word for drug
A science that studies drug effects within a living system, biochemical and physiological aspects Deals with all drugs used in society today, legal or illegal, including street, prescription, and non-prescription or over –the-counter medications

2
Drug A drug is defined as any substance; chemical agent; used in the
Diagnosis Cure Treatment prevention of a disease or condition

3
Drug Names Chemical Name Generic Name Trade Name

4
Chemical Name Describes its molecular structure and distinguishes it from other drugs

5
Generic name Determined by the pharmaceutical company along with a special organization known as the U.S. Adopted Names Council (USAN)
 .")
6
Trade Name Or brand name- the manufacturer selects alone…can become a registered trademark. They are the only one who can advertise and market the drug under that name.

7
How is the Trade Name Chosen?
The particular spelling of a brand name drug is proposed by a manufacturer for one of several reasons.

8
1. To indicate the disease process being treated
Azmacort- treats asthma Rythmol- treats cardiac arrhythmias

9
2. To simplify the generic name
Pseudoephedrine to Sudefed Haloperidol to Haldol Ciprofloxacin to Cipro

10
3. To indicate the duration
Slow-K slow release potassium supplement

11
Prescription Drugs Or legend drugs
Means in order to obtain drug, you must have a legal prescription

13
Non-Prescription Drugs
Or Over-the-Counter (OTC) drugs Drug that may be purchased without a prescription
 drugs. Drug that may be purchased without a prescription.")
14
Sources of Drugs Drugs have been identified or derived from four main sources: Plants Animals Minerals and Mineral Products Synthetic or Chemical Substances Made in the Laboratory

16
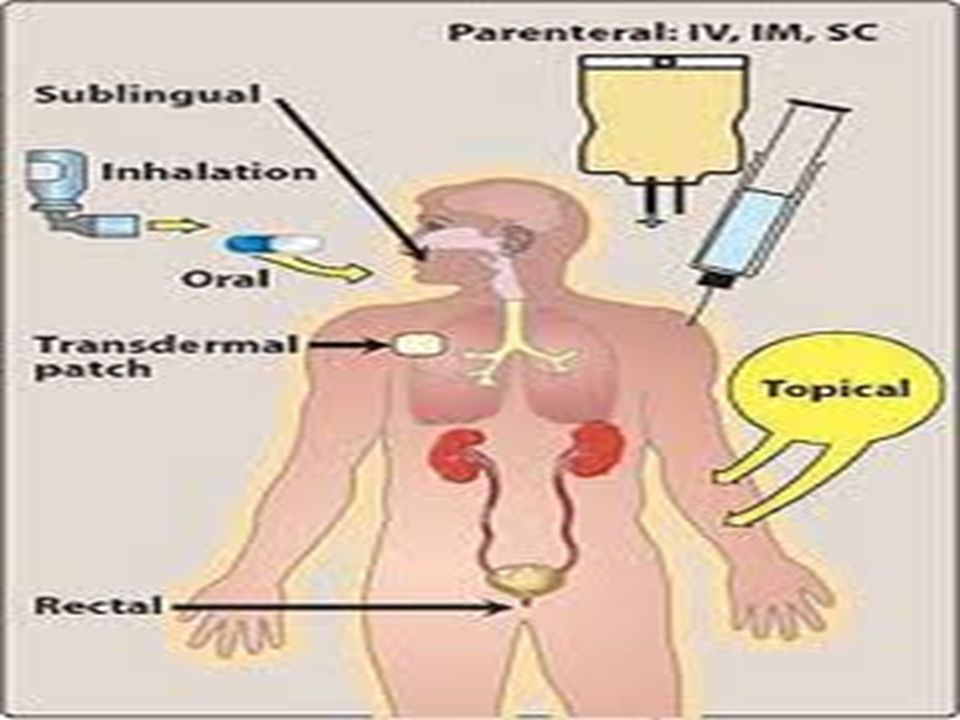
Routes of drug administration

17
Routes of Drug Administration
Important Info The route of administration (ROA) that is chosen may have a profound effect upon the speed and efficiency with which the drug acts
 that is chosen may have a profound effect upon the speed and efficiency with which the drug acts.")
19
The main routes of drug entry into the body may be divided into two classes:
Enteral Parenteral

20
Enteral Routes sublingual - placed under the tongue
Enteral - drug placed directly in the GI tract: sublingual - placed under the tongue oral - swallowing (p.o., per os) rectum - Absorption through the rectum
 rectum - Absorption through the rectum.")
21
Sublingual/Buccal Advantages
Some drugs are taken as smaller tablets which are held in the mouth or under the tongue. Advantages rapid absorption drug stability avoid first-pass effect

22
Sublingual/Buccal Disadvantages inconvenient small doses
unpleasant taste of some drugs

23
Oral Disadvantages Sometimes inefficient - only part of the drug may be absorbed First-pass effect - drugs absorbed orally are initially transported to the liver via the portal vein irritation to gastric mucosa - nausea and vomiting

24
Oral Disadvantages destruction of drugs by gastric acid and digestive juices effect too slow for emergencies unpleasant taste of some drugs unable to use in unconscious patient

25
First-pass Effect The first-pass effect is the term used for the hepatic metabolism of a pharmacological agent when it is absorbed from the gut and delivered to the liver via the portal circulation. The greater the first-pass effect, the less the agent will reach the systemic circulation when the agent is administered orally

26
First-pass Effect Magnitude of first pass hepatic effect: Extraction ratio (ER) ER = CL liver / Q ; where Q is hepatic blood flow (usually about 90 L per hour. Systemic drug bioavailability (F) may be determined from the extent of absorption (f) and the extraction ratio (ER): F = f x (1 -ER)
 may be determined from the extent of absorption (f) and the extraction ratio (ER): F = f x (1 -ER)")
27
First-pass Effect

28
RECTAL ADMINISTRATION:
Absorption across the rectal mucosa occurs by passive diffusion. This route of administration is useful in children, old people and unconscious patients. Eg., drugs that administered are: aspirin, acetaminophen, theophylline, indomethacin, promethazine & certain barbiturates. KLECOP, Nipani 08/10/2010

29
Rectal Advantages: Suitable for unconscious patients and children
2. suitable if patient is nauseous or vomiting 3. easy to terminate exposure 4. good for drugs affecting the bowel such as laxatives Disadvantages: absorption may be variable irritating drugs contraindicated

30
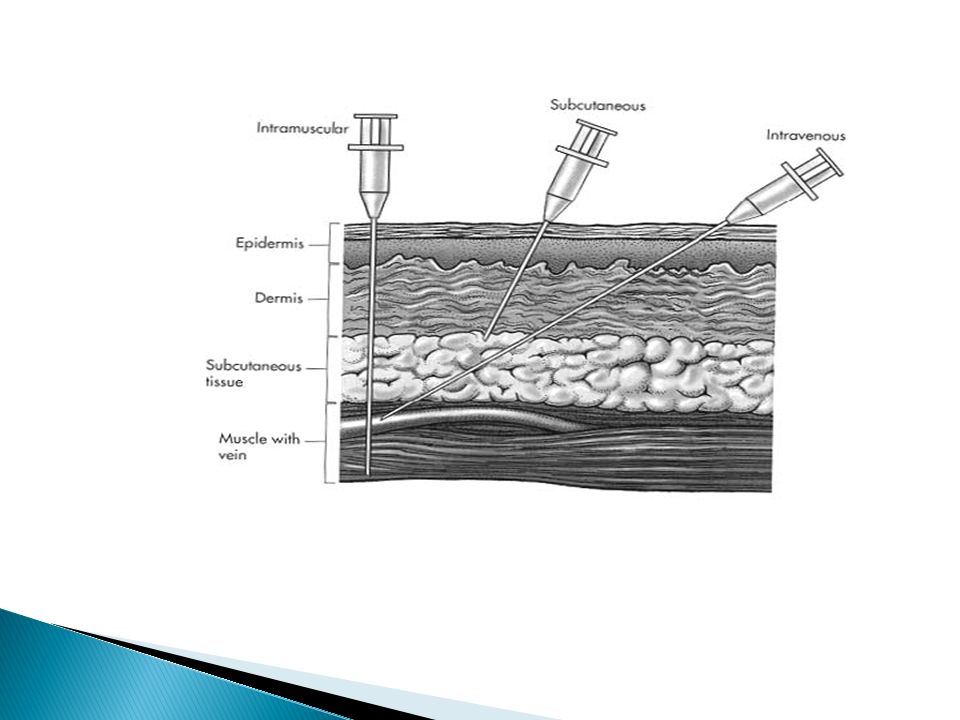
Parenteral Routes Intravascular (IV, IA)- placing a drug directly into the blood stream Intramuscular (IM) - drug injected into skeletal muscle Subcutaneous - Absorption of drugs from the subcutaneous tissues Intrathecal : into CSF

32
Intravascular Absorption phase is bypassed (100% bioavailability)
1.precise, accurate and almost immediate onset of action, 2. large quantities can be given, fairly pain free Disadvantages a-. greater risk of adverse effects b- high concentration attained rapidly C- risk of embolism

33
Intramuscular 1. very rapid absorption of drugs in aqueous solution
2. Slow release preparations Disadvantages pain at injection sites for certain drugs

34
Subcutaneous slow and constant absorption
2. absorption is limited by blood flow, affected if circulatory problems exist 3. concurrent administration of vasoconstrictor will slow absorption

35
Inhalation 1. gaseous and volatile agents and aerosols
2. rapid onset of action due to rapid access to circulation a. large surface area b. thin membranes separates alveoli from circulation c. high blood flow

36
Topical Mucosal membranes (eye drops, antiseptic) Skin
a. Dermal - rubbing in of oil or ointment (local action, sun screen, an callus removal) b. Transdermal - absorption of drug through skin (systemic action) i. stable blood levels ii. no first pass metabolism iii. drug must be potent or patch becomes too large
 b. Transdermal - absorption of drug through skin (systemic action) i. stable blood levels. ii. no first pass metabolism. iii. drug must be potent or patch becomes too large.")
37
Intra nasal administration
Drugs generally administered by intra nasal route for treatment of local condition such as perennial rhinitis, allergic rhinitis and nasal decongestion etc.

38
Route for administration
-Time until effect- intravenous seconds intraosseous seconds endotracheal 2-3 minutes inhalation 2-3 minutes sublingual 3-5 minutes intramuscular minutes subcutaneous minutes rectal 5-30 minutes ingestion minutes transdermal (topical) variable (minutes to hours)
 variable (minutes to hours)")
39
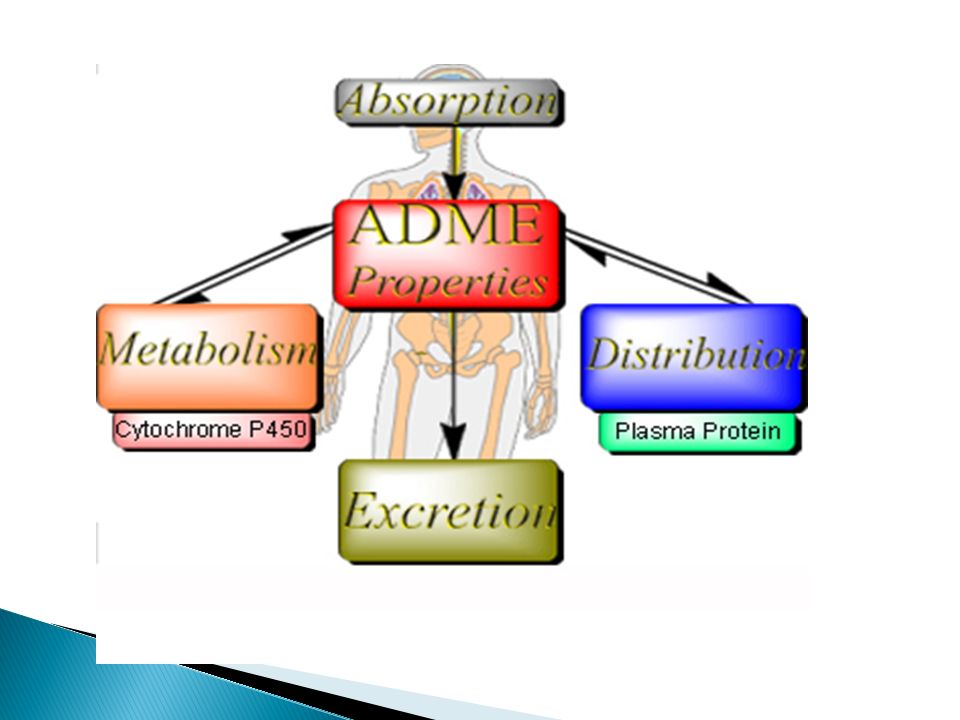
Aspects of Drug Pharmacokinetics (ADME)
Drug at site of administration Absorption Drug in plasma Distribution Drug/metabolites in tissues Metabolism Elimination Drug/metabolites in urine, feces, bile

41
Absorption Definition :
The process of movement of unchanged drug from the site of administration to systemic circulation. The ultimate goal is to have the drug reach the site of action in a concentration which produces a pharmacological effect. No matter how the drug is given (other than IV) it must pass through a number of biological membranes before it reaches the site of action.
 it must pass through a number of biological membranes before it reaches the site of action.")
42
LIPID BILAYER KLECOP, Nipani 08/10/2010

43
DIFFUSION THROUGH MEMBRANES
the Rate dependent on polarity and size. Polarity estimates partition coefficient. The greater the lipid solubility – the faster the rate of diffusion Smaller molecules penetrate more rapidly. Highly permeable to O2, CO2, NO and H2O . Large polar molecules – sugar, amino acids, phosphorylated intermediates – poor permeability These are essential for cell function – must be actively transported

44
MOVEMENT OF SUBSTANCES ACROSS CELL MEMBRANES
KLECOP, Niani

45
MECHANISMs OF DRUG ABSORPTION
Passive diffusion Carrier- mediated transport a) Facilitated diffusion b) Active transport PINOCYTOSIS KLECOP, Nipani 08/10/2010
 Facilitated diffusion. b) Active transport. PINOCYTOSIS. KLECOP, Nipani. 08/10/2010.")
46
PASSIVE DIFFUSION Also known as non- ionic diffusion.
It depends on the difference in the drug concentration on either side of the membrane. Absorption of 90% of drugs. The driving force for this process is the concentration or electrochemical gradient.

47
2) CARRIER MEDIATED TRANSPORT MECHANISM
Involves a carrier (a component of the membrane) which binds reversibly with the solute molecules to be transported to yield the carrier solute complex which transverses across the membrane to the other side where it dissociates to yield the solute molecule The carrier then returns to its original site to accept a fresh molecule of solute. There are two types of carrier mediated transport system: a) facilitated diffusion b) active transport
 which binds reversibly with the solute molecules to be transported to yield the carrier solute complex which transverses across the membrane to the other side where it dissociates to yield the solute molecule. The carrier then returns to its original site to accept a fresh molecule of solute. There are two types of carrier mediated transport system: a) facilitated diffusion. b) active transport.")
48
a) Facilitated diffusion
This mechanism driving force is concentration gradient. In this system, no use of energy is involved (down-hill transport), therefore the process is not inhibited by metabolic poisons that interfere with energy production.
, therefore the process is not inhibited by metabolic poisons that interfere with energy production.")
49
b) Active transport More important process than facilitated diffusion.
The driving force is against the concentration gradient or uphill transport. Since the process is uphill, energy is required in the work done by the barrier. As the process requires energy, it can be inhibited by metabolic poisons that interfere with energy production.

50
Drug Absorption Active vs. Passive
Active transport: Carrier-mediated Energy-dependent Against conc gradient Shows carrier saturation kinetics Passive transport Energy-independent No carrier involved Along conc gradient No saturation kinetics ATP Carrier-mediated energy-dependent active transport ADP + Pi Passive diffusion of a water-sol drug via aqueous channel AH B Passive diffusion of a lipid-sol drug A- BH+

51
3) Pinocytosis This process is important in the absorption of oil soluble vitamins & in the uptake of nutrients.
 Pinocytosis This process is important in the absorption of oil soluble vitamins & in the uptake of nutrients.")
52
PHYSICOCHEMICAL FACTORS
Drug transported by passive diffusion depend upon: dissociation constant, pKa of the drug lipid solubility, K o/w pH at absorption site. Most drugs are either weak acids or weak bases whose degree of ionization is depend upon pH of biological fluid.

53
For a drug to be absorbed, it should be unionized and the unionized portion should be lipid soluble. Only non-ionized fraction of drugs (acids or bases is absorbed The fraction of drug remaining unionized is a function of both Dissociation constant (pKa) and pH of solution.
 and pH of solution.")
54
HENDERSON HASSELBATCH EQUATION For acid, pKa - pH = log[ Cu/Ci ]
For base, pKa – pH = log[ Ci/Cu ] Eg. Weak acid aspirin (pKa=3.5) in stomach (pH=1) will have > 99%of unionized form so gets absorbed in stomach Weak base quinine (pKa=8.5) will have very negligible unionization in gastric pH so negligible absorption Several prodrugs have been developed which are lipid soluble to overcome poor oral absorption of their parent compounds. KLECOP, Nipani 08/10/2010
![HENDERSON HASSELBATCH EQUATION For acid, pKa - pH = log[ Cu/Ci ]](http://slideplayer.com/slide/7016140/24/images/54/HENDERSON+HASSELBATCH+EQUATION+For+acid%2C+pKa+-+pH+%3D+log%5B+Cu%2FCi+%5D.jpg "For base, pKa – pH = log[ Ci/Cu ] Eg. Weak acid aspirin (pKa=3.5) in stomach (pH=1) will have > 99%of unionized form so gets absorbed in stomach. Weak base quinine (pKa=8.5) will have very negligible unionization in gastric pH so negligible absorption. Several prodrugs have been developed which are lipid soluble to overcome poor oral absorption of their parent compounds. KLECOP, Nipani. 08/10/2010.")
55
Factors Affecting GIT Absorption
Blood Flow To Absorptive Site: Greater blood flow raises absorption Intestine has greater BF than stomach Total Surface Area of Absorptive Site: Intestinal microvilli increases surface area to fold that of the stomach favoring intestinal absorption Contact Time at Absorptive Site: Diarrhea reduces absorption Accelerated gastric emptying→ faster delivery to intestinal large surface → increased absorption

56
Factors Affecting GIT Absorption
Food: Presence of food in the gut reduces/delays drug absorption from GIT Increased splanchnic blood flow during eating increases drug absorption Ionized drugs as tetracycline can form insoluble complexes with Ca2+ in food/milk. Formulation Factors: Solid dosage forms dissolution & solubility are essential Aqueous solutions are absorbed more quickly than tablets or suspensions

57
Factors affecting absorption from GIT
Stomach: The surface area for absorption of drugs is relatively small in the stomach due to the absence of macrovilli & microvilli. Extent of drug absorption is affected by variation in the time it takes the stomach to empty, i.e., how long the dosage form is able to reside in stomach. Drugs which are acid labile must not be in contact with the acidic environment of the stomach

58
PHYSIOLOGICAL FACTORS:
Gastrointestinal (Gi) Physiology Influence Of Drug Pka And Gi Ph On Drug Absorbtion Git Blood Flow Gastric Emptying………………..contact time Disease States Total surface area
 Physiology. Influence Of Drug Pka And Gi Ph On Drug Absorbtion. Git Blood Flow. Gastric Emptying………………..contact time. Disease States. Total surface area.")
59
Intestine Major site for absorption of most drugs due to its large surface area (0.33 m2 ). It is 7 meters in length and is approximately cm in diameter. These folds possess finger like projections called Villi which increase the surface area 30 times ( 10 m2). From the surface of villi protrude several microvilli which increase the surface area 600 times ( 200 m2). Blood flow is times that of stomach. PH Range is 5–7.5 , favorable for most drugs to remain unionized. Peristaltic movement is slow, while transit time is long. Permeability is high. All these factors make intestine the best site for absorption of most drugs.
. From the surface of villi protrude several microvilli which increase the surface area 600 times ( 200 m2). Blood flow is 6-11 times that of stomach. PH Range is 5–7.5 , favorable for most drugs to remain unionized. Peristaltic movement is slow, while transit time is long. Permeability is high. All these factors make intestine the best site for absorption of most drugs.")
60
Large intestine : The major function of large intestine is to absorb water from ingestible food residues which are delivered to the large intestine in a fluid state, & eliminate them from the body as semi solid feces. Only a few drugs are absorbed in this region.

61
Bioavailability the proportion of the drug in a dosage form available to the body i.v injection gives 100% bioavailability.

62
BIOAVAILABIITY Time Injected Dose Serum Concentration Oral Dose
Fraction of a drug reaching systemic circulation in chemically unchanged form after a particular route First pass metabolism, i.e., rapid hepatic metabolism, reduces bioav. (lidocaine, propranolol, nitrates) Drug solubility Chemical instability in gastric pH (penicillin G, insulin) Drug formulation: Standard & SR formulations Bio = AUC oral/AUC IV x 100 Injected Dose Serum Concentration Oral Dose Time
 Drug solubility. Chemical instability in gastric pH (penicillin G, insulin) Drug formulation: Standard & SR formulations. Bio = AUC oral/AUC IV x 100. Injected Dose. Serum Concentration. Oral Dose. Time.")
63
DISTRIBUTION The body is a container in which a drug is distributed by blood (different flow to different organs) - but the body is not homogeneous. Factors affecting drug delivery from the plasma: A- blood flow: kidney and liver higher than skeletal muscles and adipose tissues. B- capillary permeability: 1- capillary structure: blood brain barrier 2- drug structure C- binding of drugs to plasma proteins and tissue proteins
 - but the body is not homogeneous. Factors affecting drug delivery from the plasma: A- blood flow: kidney and liver higher than skeletal muscles and adipose tissues. B- capillary permeability: 1- capillary structure: blood brain barrier. 2- drug structure. C- binding of drugs to plasma proteins and tissue proteins.")
72
DEFINITION OF PHARMACOKINETICS AND PHARMACODYNAMICS

74
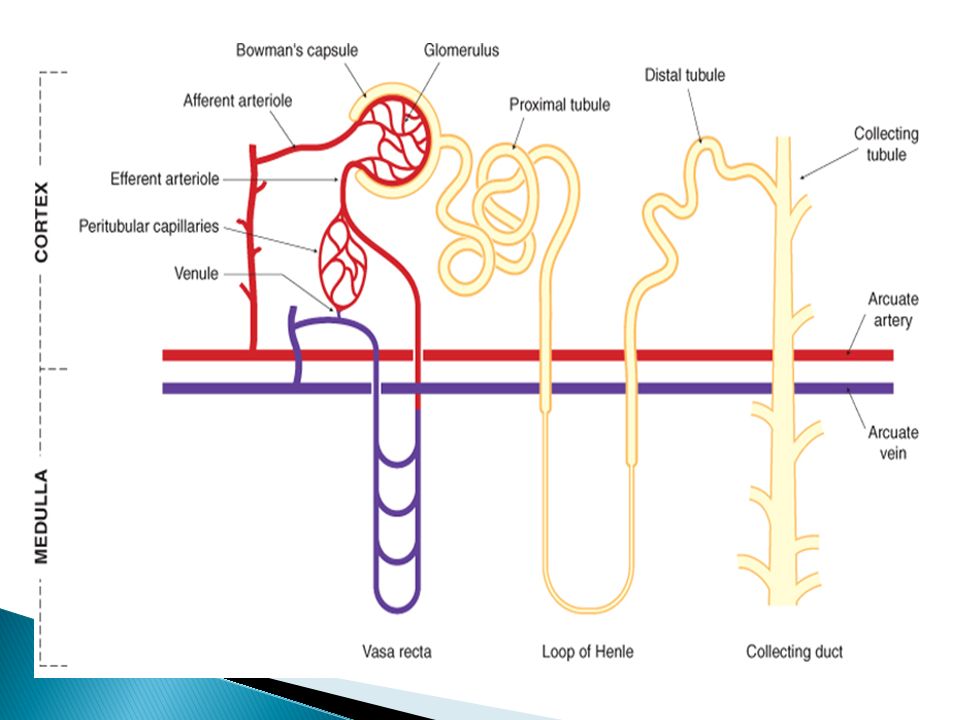
Renal Excretion Glomerular filtration depends on:
Renal blood flow & GFR; direct relationship Plasma protein binding; only free unbound drugs are filtered Tubular Secretion in the proximal renal tubule mediates raising drug concentration in PCT lumen Organic anionic & cationic transporters (OAT & OCT) mediate active secretion of anionic & cationic drugs Passive diffusion of uncharged drugs Facilitated diffusion of charged & uncharged drugs Penicillin is an example of actively secreted drugs
 mediate active secretion of anionic & cationic drugs. Passive diffusion of uncharged drugs. Facilitated diffusion of charged & uncharged drugs. Penicillin is an example of actively secreted drugs.")
75
Renal Excretion Tubular re-absorption in DCT:
Because of water re-absorption, urinary D concentration increases towards DCT favoring passive diffusion of un- ionized lipophillic drugs It leads to lowering urinary drug concentration Urinary pH trapping: Chemical adjustment of urinary pH can inhibit or enhance tubular drug reabsorption For example, aspirin overdose can be treated by urine alkalinization with Na Bicarbonate (ion trapping) and increasing urine flow rate (dilution of tubular drug concentration) Ammonium chloride can be used as urine acidifier for basic drug overdose treatment
 and increasing urine flow rate (dilution of tubular drug concentration) Ammonium chloride can be used as urine acidifier for basic drug overdose treatment.")
76
Drug Elimination Pulmonary excretion of drugs into expired air:
Gases & volatile substances are excreted by this route No specialized transporters are involved Simple diffusion across cell membrane predominates. It depends on: Drug solubility in blood: more soluble gases are slowly excreted Cardiac output rise enhance removal of gaseous drugs Respiratory rate is of importance for gases of high blood solubility Biliary excretion of few drugs into feces Such drugs are secreted from the liver into the bile by active transporters, and then into duodenum Examples: digoxin, steroid hormones, some anticancer agents Some drugs undergo enterohepatic circulation back into systemic circulation

77
CLEARANCE:- Is defined as the hypothetical volume of body fluids containing drug from which the drug is removed/ cleared completely in a specific period of time. Expressed in ml/min. CL = kVD, k: elimination rate constant KLECOP, Nipani

78
Clearance It is ability of kidney, liver and other organs to eliminate drug from the bloodstream Units are in L/hr or L/hr/kg Used in determination of maintenance doses Drug metabolism and excretion are often referred to collectively as clearance The endpoint is reduction of drug plasma level Hepatic, renal and cardiac failure can each reduce drug clearance and hence increase elimination T1/2 of the drug

79
TOTAL BODY CLEARANCE:-
Is defined as the sum of individual clearances by all eliminating organs is called total body clearance/ total systemic clearance. Total Body Clearance = CLliver + CLkidney + CLlungs +CLx KLECOP, Nipani

80
Metabolism & Excretion Kinetics
Elimination (metabolism + excretion) of most drugs follow first-order kinetics at therapeutic dose level Amount of drug cleared in a given unit of time is directly proportional to the concentration of the drug according to Michaelis-Menten (linear) kinetics: Only few drugs (e.g., phenytoin, alcohol) show saturation clearance (Zero-order, non-linear) kinetics Clearance mechanisms become saturated at therapeutic level, and clearance remain constant even with increased drug plasma level SLOW ELIMINATION at therapeutic levels leads to toxic reactions E = Vmax x C km + C
 of most drugs follow first-order kinetics at therapeutic dose level. Amount of drug cleared in a given unit of time is directly proportional to the concentration of the drug according to Michaelis-Menten (linear) kinetics: Only few drugs (e.g., phenytoin, alcohol) show. saturation clearance (Zero-order, non-linear) kinetics. Clearance mechanisms become saturated at therapeutic level, and clearance remain constant even with increased drug plasma level. SLOW ELIMINATION at therapeutic levels leads. to toxic reactions. E = Vmax x C. km + C.")
81
Therapeutic success of a rapidly & completely absorbed drug.
Not only the magnitude of drug that comes into the systemic circulation but also the rate at which it is absorbed is important this is clear from the figure. Plasma Drug Conc. Minimum effective conc. Therapeutic failure of a slowly absorbed drug. Subtherapeutic level Time

82
Loading Dose = Target Plasma C x VD
What Is the is the loading dose required fro drug A if: target concentration is 30 mg/L VD is 0.75 L/kg, patients weight is 75 kg Answer VD = 0.75 L/kg x 75 kg = L Target Conc. = 10 mg/L Dose = 30 mg/L x L = mg

83
= CL x target steady state drug concentration
Maintenance Dose Maintenance Dose = CL x target steady state drug concentration The units of CL are in L/hr or L/hr/kg Maintenance dose will be in mg/hr

84
Pharmacodynamics (how drugs work on the body)
It is the study of biochemical and physiological effects of drugs and their mechanism of action at organ level as well as cellular level.

85
LONGITUDNAL SECTION OF KIDNEY
KLECOP, Nipani

86
Half-life (t1/2) Half-life: is a derived parameter, completely determined by volume of distribution and clearance. (Units = time) As Vd increases t1/2 increases
 As Vd increases t1/2 increases.")
87
Steady-State Steady-state occurs after a drug has been given for approximately 4-5 t1/2 At steady-state the rate of drug administration equals the rate of elimination Plasma concentration after each dose is approximately the same

88
Importance of Steady State (SS)
At SS Rate in = Rate Out Steady state is reached usually within 4 – 5 half-lives at linear kinetics It is important for drug concentrations interpretation in: Therapeutic Drug Monitoring (TDM) Evaluation of clinical response
 Evaluation of clinical response.")
89
Dosing and Steady State
Dosing: Administration of medication over time, so that therapeutic levels can be achieved. Steady-state: drug accumulates and plateaus at a particular level rate of accumulation determined by half life reach steady state in about five times the elimination half-life

90
How Drugs Act: Pharmacodynamics

91
Pharmacodynamics (how drugs work on the body)
It is the study of biochemical and physiological effects of drugs and their mechanism of action at organ level as well as cellular level.

92
Principles of drug action
Do NOT impart new functions on any system, organ or cell Only alter the PACE of ongoing activity STIMULATION DEPRESSION REPLACEMENT CYTOTOXIC ACTION

93
Targets of drug action Majority of drugs interact with target biomolecules: Usually a Protein ENZYMES ION CHANNELS TRANSPORTERS RECEPTORS

94
local anesthetic & amiloride
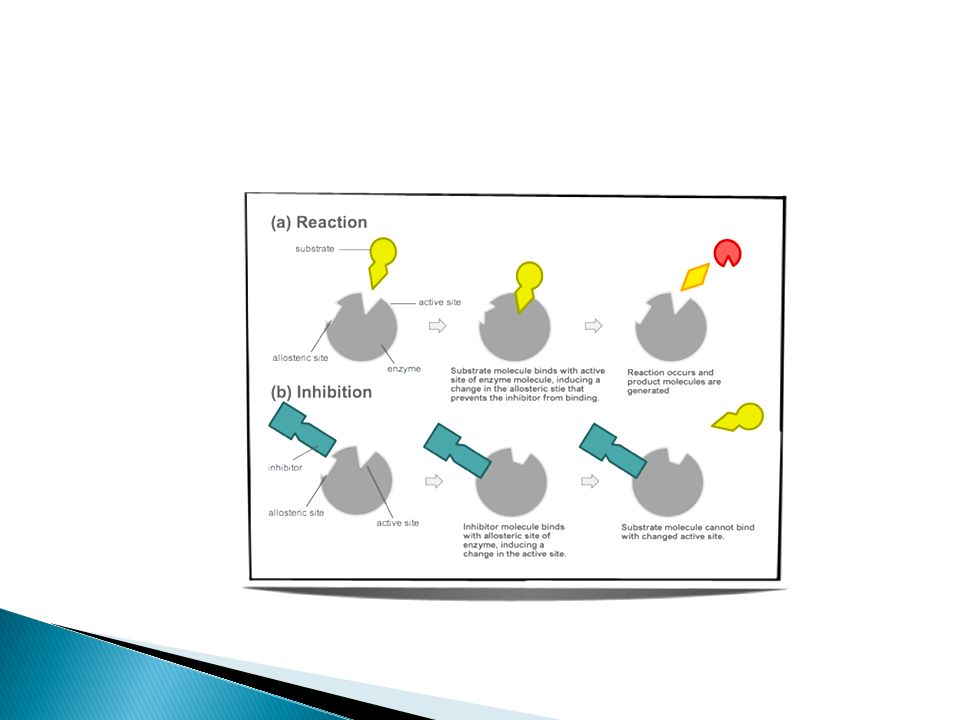
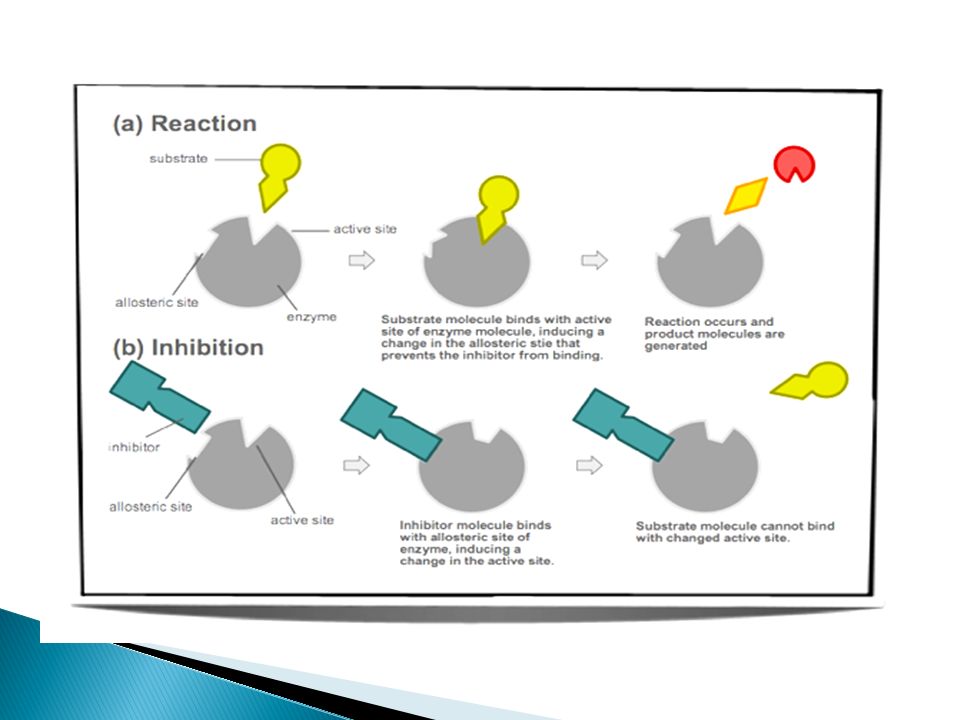
I- Ion Channels Direct Physical blocking of channel local anesthetic & amiloride Modulator Bind to the channel protein itself Ca channel blockers

95
II- Enzymes ChE inhibitors α-Methyl dopa

96
II- Enzymes (cont.) Drug acts as
Substrate leading to reversible OR irreversible inhibition of enzyme reversible inhibition of cholinesterase by neostigmine Irreversible inhibition of cyclo-oxygenase by aspirin True/False substrate L-DOPA converted into dopamine α-methyldopa converted into α- methylnorepinephrine (false transmitter)
")
97
III- Carrier Molecules
What is carrier molecule? Carrier protein molecules function to transport ions & small organic molecules (too polar to penetrate) across cell membranes.
 across cell membranes.")
98
III- Carrier Molecules
They possess a recognition site that confers specificity for a particular carried agent. Such recognition sites can be targets for drugs where they block the transport system. An example is the inhibition of cardiac Na+K+-ATPase by cardiac glycosides.

99
IV- Receptors cellular macromolecular proteins located either in the cell membrane or less frequently in the cytoplasm. Definition: It is defined as a macromolecule or binding site located on cell surface or inside the effector cell that serves to recognize the signal molecule/drug and initiate the response to it, but itself has no other function, e.g. G- protein coupled receptor.

100
They have specific recognition sites that bind selectively with a structurally-related group of synthetic drugs and endogenous mediators (ligands). They responsible for transducing extracellular signals into intracellular response

101
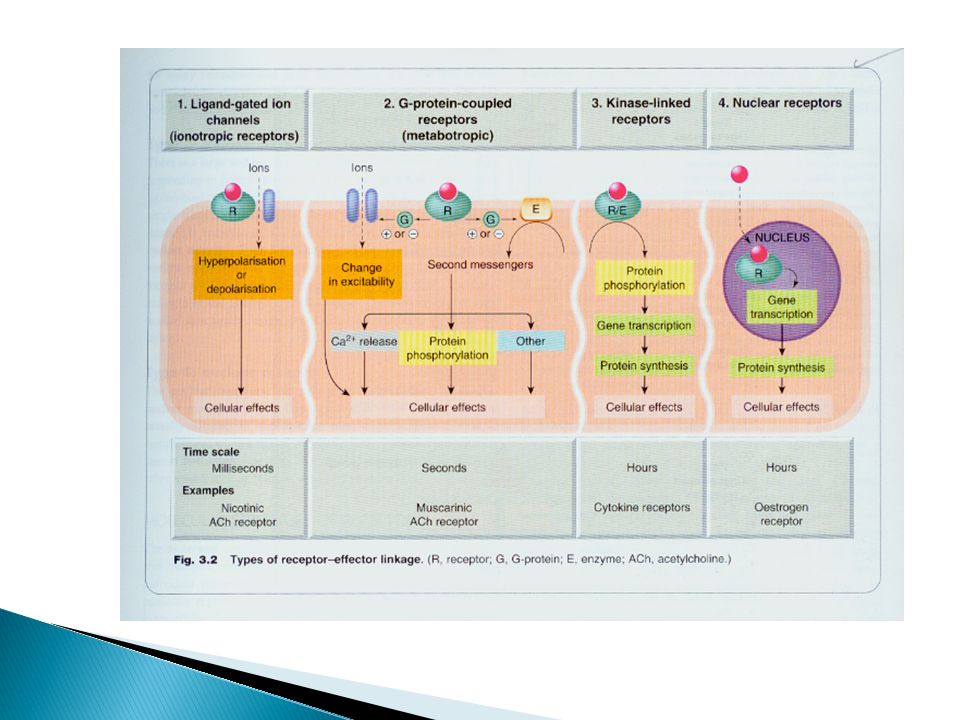
IV- Receptors There are FOUR types of receptors, classified according to their molecular structure and the nature of the receptor-effector linkage

103
Receptor structure Membrane receptors are usually composed of three parts: more than one hydrophobic membrane- spanning α-helical segment the extracellular ligand-binding domain the intracellular transduction domain

105
Ligand-gated ion channel (inotropic)
They are responsible for regulation of ions across cell membrane Binding of ligand to receptor → opening or closure of channel Response is very rapid (few milisec) E.g. Nicotinic receptors in the skeletal muscle δ-aminobuteric acid (GABA) receptors in the brain
 E.g. Nicotinic receptors in the skeletal muscle. δ-aminobuteric acid (GABA) receptors in the brain.")
106
nAch receptor: pentamer protein (α2βγδ),
,")
107
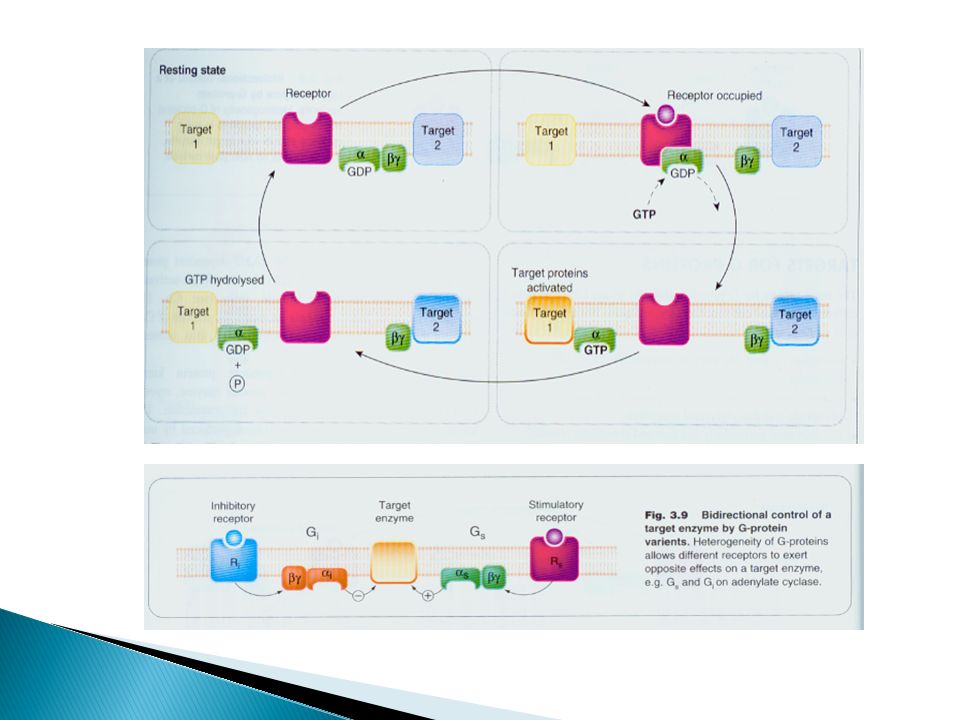
Class 2: G-protein-Coupled (Metabotropic) Receptors
Membrane bound receptors which are bound to effector system through G- proteins. These are hetero trimeric molecules having 3 subunits α,β and ϒ. Based on α-sub unit they are further classified into 3 main varieties Gs, Gi and Gq G-protein controls the activity of an effector protein; a membrane enzyme or an ion channel Activation/inhibition of the effector enzyme increase/decrease the release of a diffusible second messenger such as cAMP or IP3

108
Subtypes of G-proteins - Targets (Second messenger systems)
Ion chanels: Na+ / H+ exchange Enzyms: Gi Inhib. Adenylyl cyclase Gs Stimul. Adenylyl cyclase Gq Stimul. Phospholipase C One ligand can bind to more than one type of G-proteins coupled receptors second messenger pathways

110
2nd Messenger Phospholipase C Adenylate cyclase IP3 cAMP DAG Activation of protein Kinase C Regulation of free Ca in the cell

111
Enzyme linked receptors
They have cytosolic enzyme in their structure. Binding of a ligand to extracellular domain activate or inhibit the enzyme. Duration of response is minutes to hrs. They are two main groups Tyrosine-kinase-linked receptors such as receptors for insulin, growth factors and many cytokines, Guanylate cyclase-coupled receptors for atrial natriuretic peptide (ANP)
")
112
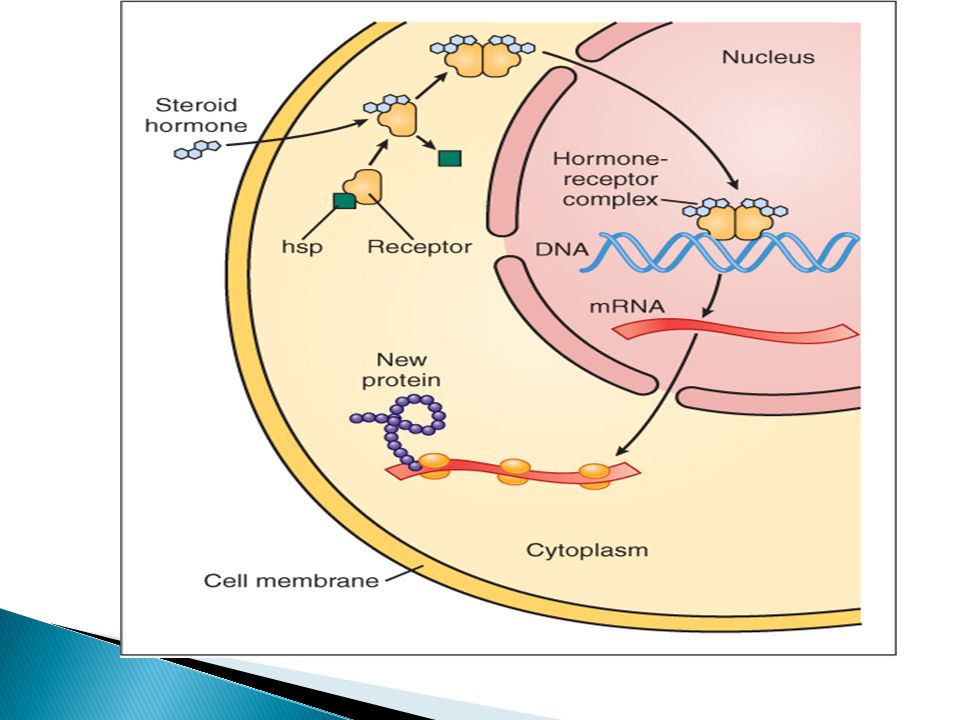
Class 4: Gene Transcription-Regulating Receptor
This is the only intracellular cytoplasmic protein receptors, NO membrane segments. The drug should diffuse into the cell to interact with receptor i.e. the drug should be lipid soluble. E.g. Steroid & thyroid hormones. It takes time for onset of action i.e. time for protein synthesis and longer duration of action (hrs to days)
")
114
Drug-Receptor Interactions
Drug (D) + Receptor (R) K K2 DR complex Pharmacologic Response
 + Receptor (R) K1 K2. DR complex. Pharmacologic Response.")
115
Lock and key theory

116
Drugs binding to the receptors is governed by Law of Mass Action.
The number of receptors [R] occupied by a drug depends on the drug concentration [D] and the drug-receptor association and dissociation rate constants (K1 & K2). Affinity: Ability of a substrate to bind with receptor Intrinsic activity (IA): Capacity to induce functional change in the receptor
. Affinity: Ability of a substrate to bind with receptor. Intrinsic activity (IA): Capacity to induce functional change in the receptor.")
117
Affinity & Efficacy Key & Lock theory Affinity DR complex Drug Receptor Efficacy Affinity is the tendency of drug to combine with its receptor Efficacy is the ability of a drug to initiate a cellular effect Biological response

118
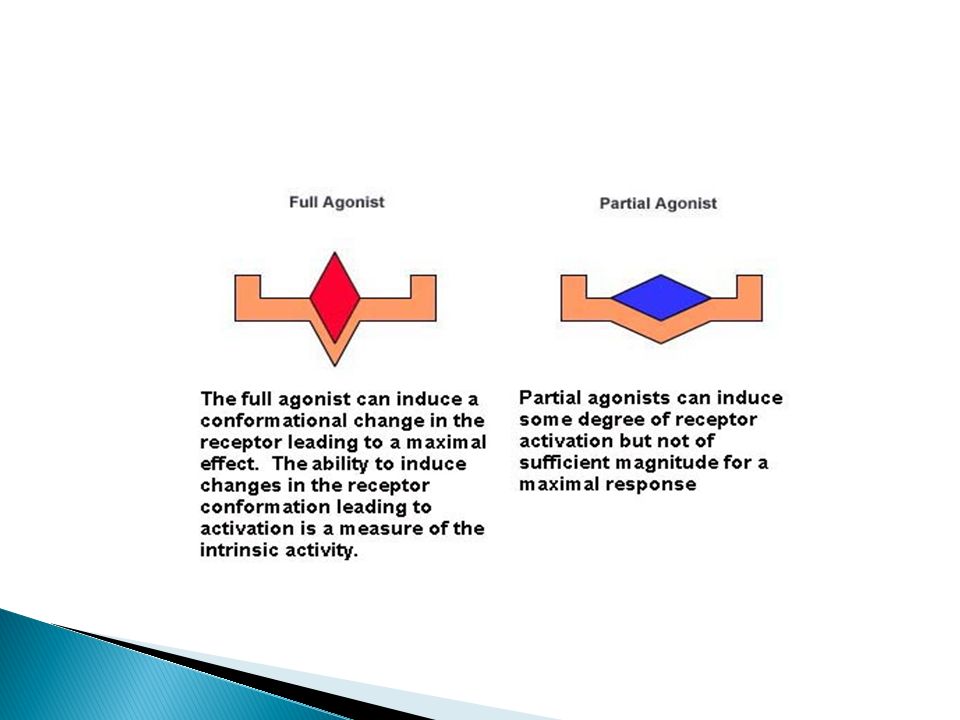
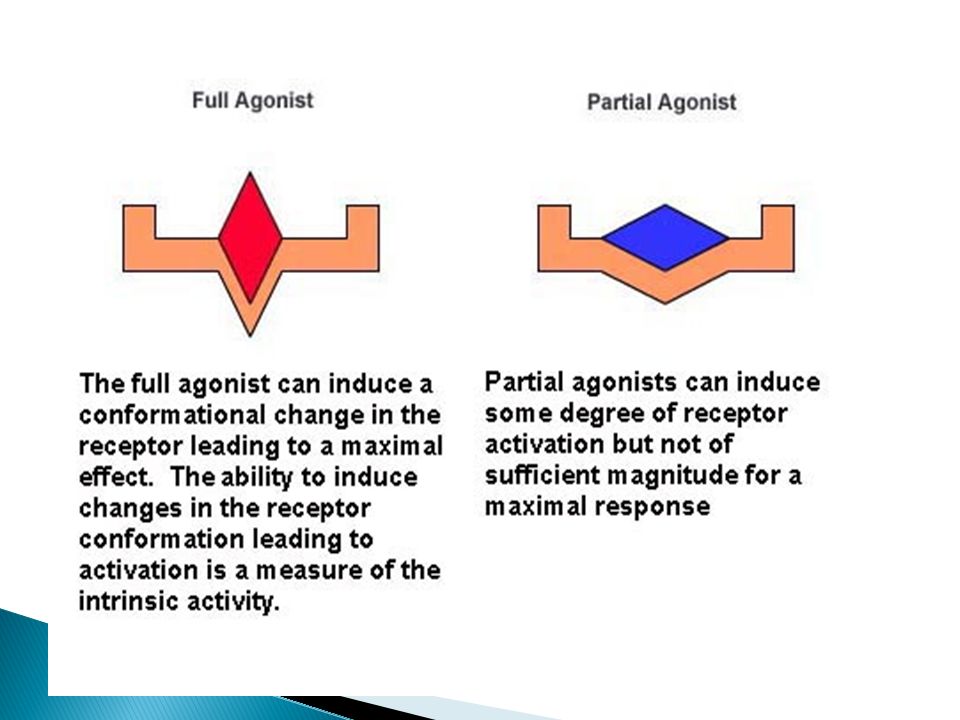
According to Efficacy, the drug may be
Agonist An agent which activates a receptor to produce an effect similar to a that of the physiological signal molecule, e.g. Muscarine and Nicotine) response. it has affinity & intrinsic activity i.e. the drug binds to a receptor and produce biological response like endogenous ligand. Antagonist an agent which prevents the action of an agonist on a receptor or the subsequent response, but does not have an effect of its own, e.g. atropine and muscarine no response. It has affinity but without intrinsic activity Efficacy = zero
 response. it has affinity & intrinsic activity i.e. the drug binds to a receptor and produce biological response like endogenous ligand. Antagonist. an agent which prevents the action of an agonist on a receptor or the subsequent response, but does not have an effect of its own, e.g. atropine and muscarine no response. It has affinity but without intrinsic activity. Efficacy = zero.")
119
Partial agonist or antagonist
It has efficacy > zero but < full agonist, even if all receptors are occupied It has affinity greater, less or the same as full agonist It decrease the response of the agonis. Inverse agonist: an agent which activates receptors to produce an effect in the opposite direction to that of the agonist, e.g. DMCM on bzp receptors opposite response Ligand: any molecule which attaches selectively to particular receptors or sites (only binding or affinity) If explained Agonist: Affinity+ IA Antagonist: Affinity+ IA (0) Partial agonist: Affinity + IA (0 to 1) Inverse agonist: Affinity + IA (0 to -1)
 If explained. Agonist: Affinity+ IA. Antagonist: Affinity+ IA (0) Partial agonist: Affinity + IA (0 to 1) Inverse agonist: Affinity + IA (0 to -1)")
121
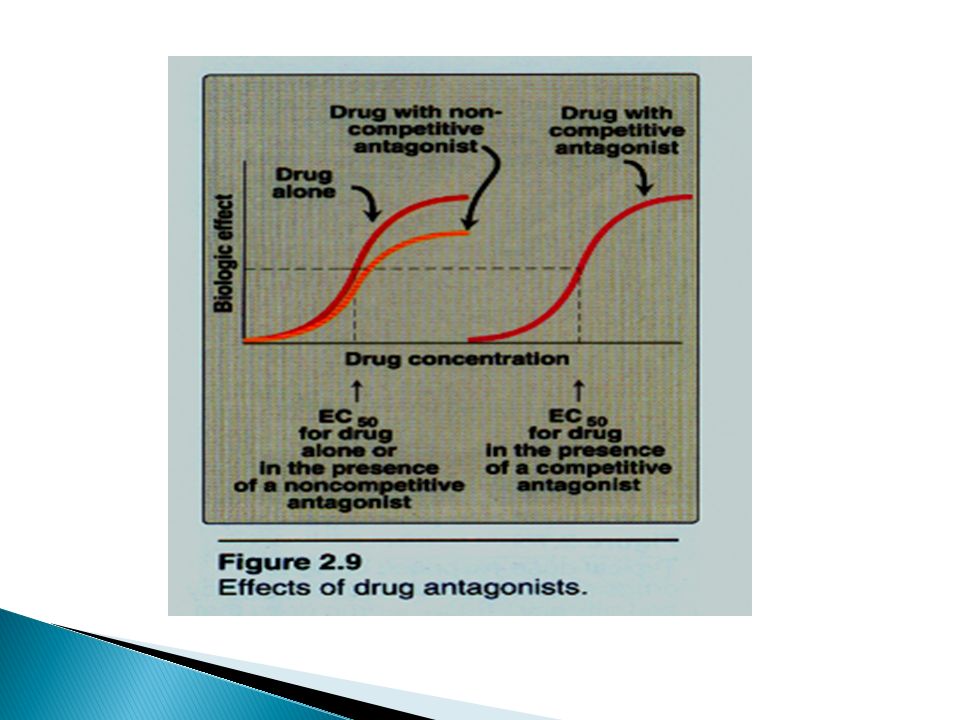
Types of drug antagonist at the receptor
Competitive antagonism Ag & Antag have the same binding site on the receptor. Increasing agonist concentration can restore the agonist occupancy and hence the response. They increase the ED50 of the agonist, but not Emax or the slope. e.g. NA & prazocin on α1 receptor Non competitive antagonism = allosteric Ag & Antag have different site of binding. Increasing the agonist does not affect antagonist occupancy or the receptor blockade. They cause a reduction of the slope & the max of the agonist concentration-response curve

124
Drug Effectiveness Dose-response (DR) curve: Depicts the relation between drug dose and magnitude of drug effect Drugs can have more than one effect Drugs vary in effectiveness Different sites of action Different affinities for receptors The effectiveness of a drug is considered relative to its safety (therapeutic index)
")
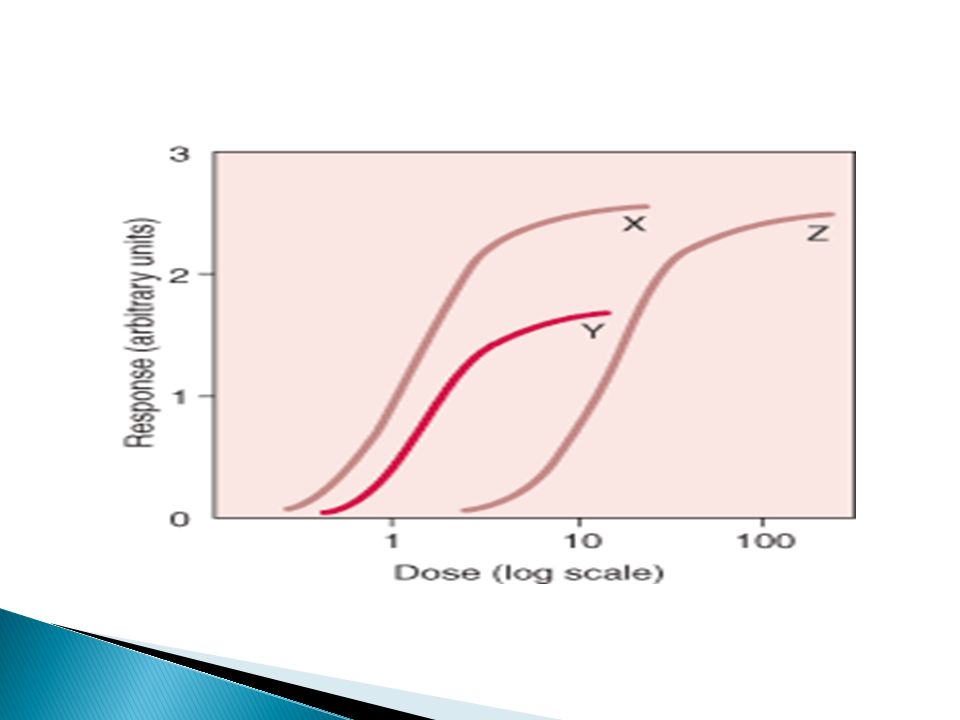
125
Dose-Response Curves By raising the dose above the “threshold dose level”, a gradual increase in response occurs. Thus, DR of similarly active drugs produce parallel DR curves, enabling us to compare between the potencies of qualitatively similar drugs.

127
Potency Amount of the drug necessary to produce certain magnitude of the effect e.g. 50% of the max. effect (EC50)
 .")
129
Efficacy = Intrinsic activity
Efficacy: the maximum effect of a drug Depends on: No. of complex formed Efficiency of coupling between the complex and the biological response (Emax) Greater efficacy is more important therapeutically
 Greater efficacy is more important therapeutically.")
132
Dose-Effect Curves

134
Enhancement of drug effects
A) Additive: 1+1= 2 B) Synergism: 1+1= 4 C) Potentiation: 1+0= 2
 Additive: 1+1= 2. B) Synergism: 1+1= 4. C) Potentiation: 1+0= 2.")
135
Therapeutic index TD50/ED50 2- Standard safety margin (SSM):
The ratio of the dose that produce toxicity to the dose that produce effective response It is obtained from quantal DRC (all-or-none effect) TD50/ED50 TD50 = The drug dose that produce toxic effect in 50% of population ED50 = The drug dose that produces a therapeutic or desired response in 50% of population Examples Warfarin (narrow index) Penicillin (wide index) 2- Standard safety margin (SSM): SSM= LD x 100 ED99
 TD50/ED50. TD50 = The drug dose that produce toxic effect in 50% of population. ED50 = The drug dose that produces a therapeutic or desired response in 50% of population. Examples. Warfarin (narrow index) Penicillin (wide index) 2- Standard safety margin (SSM): SSM= LD1 - 1 x 100. ED99.")
136
Therapeutic Index

137
Targets for drug action
1- Extracellular Sites of Drug Action Stomach: neutralize acid with base (antacids) Blood: bind metals (chelation) like lead with EDTA GI Tract: bind drugs (adsorption) with Cholestyramine. GI Tract: increase water by osmotic effects (laxatives) Kidney: increase water elimination (osmotic diuretics)
 Blood: bind metals (chelation) like lead with EDTA. GI Tract: bind drugs (adsorption) with Cholestyramine. GI Tract: increase water by osmotic effects (laxatives) Kidney: increase water elimination (osmotic diuretics)")
138
Adverse effects of drugs
Allergy: antigen-antibody………unpredictable Idiosyncrasy: genetic abnormality…….. Unpredictable Side effects: unavoidable, undesirable, normal actions by therapeutic doses. Over-dose: high dose of drugs Supersenstivity: exaggerated response to normal dose due to upregulation of receptors. Dependance: habituation and addiction.

139
Desensitization and Tachyphylaxis
When it is developing in the course of few minutes. Tolerance To describe a more gradual loss of drug-induced clinical effects that develops in the course of days or weeks. Refractoriness Used to indicate the loss of therapeutic response. Drug resistance Describes the loss of the effect of antitumor and antimicrobial drugs

140
Mechanisms of Desensitization
Receptor phosphorylation Usually by phosphorylating serine or threonine residues in the C-terminal domain of GPCRs leading to reduce efficiency and alter their binding affinity. Down-regulation of receptors Phosphorylation also signals the cell to internalize the membrane receptor leading to decrease the number of receptors on the cell membrane. In contrast, continuous or repeated exposure to antagonists initially can increase the response of the receptor (supersensitivity or up-regulation)
")
141
Mechanisms of Desensitization
Receptor phosphorylation Usually by phosphorylating serine or threonine residues in the C-terminal domain of GPCRs leading to reduce efficiency and alter their binding affinity. Down-regulation of receptors Phosphorylation also signals the cell to internalize the membrane receptor leading to decrease the number of receptors on the cell membrane. In contrast, continuous or repeated exposure to antagonists initially can increase the response of the receptor (supersensitivity or up-regulation)
")
142
Mechanisms of Desensitization
Depletion of mediators Drugs acting indirectly via transmitter release can cause depletion of that transmitter and hence loss of action e.g. amphetamine or ephedrine act by releasing catecholamines from nerve terminals. Pharmacokinetic desensitization Drugs which stimulate hepatic metabolism may enhance their own metabolism and hence a lower plasma concentration with repeated administration of the same dose e.g. barbiturates Pumping of drugs out from intracellular site (chemotherapy)
")
143
MCQs

144
MCQs The description of molecular events initiated with the ligand binding and ending with a physiologic effect is called (A) receptor down-regulation (B) signal transduction pathway (C) ligand-receptor binding (D) law of mass action (E) intrinsic activity or efficacy
 receptor down-regulation. (B) signal transduction pathway. (C) ligand-receptor binding. (D) law of mass action. (E) intrinsic activity or efficacy.")
145
MCQs The description of molecular events initiated with the ligand binding and ending with a physiologic effect is called (A) receptor down-regulation (B) signal transduction pathway (C) ligand-receptor binding (D) law of mass action (E) intrinsic activity or efficacy
 receptor down-regulation. (B) signal transduction pathway. (C) ligand-receptor binding. (D) law of mass action. (E) intrinsic activity or efficacy.")
146
MCQs A partial agonist is best described as an agent that ---- (A) has low potency but high efficacy (B) acts as both an agonist and antagonist (C) interacts with more than one receptor type (D) cannot produce the full effect, even at high doses (E) blocks the effect of the antagonist THANK YOU
 acts as both an agonist and antagonist. (C) interacts with more than one receptor type. (D) cannot produce the full effect, even at high doses. (E) blocks the effect of the antagonist. THANK YOU.")
147
Principles of drug action
Do NOT impart new functions on any system, organ or cell Only alter the PACE of ongoing activity STIMULATION DEPRESSION REPLACEMENT CYTOTOXIC ACTION

148
Targets of drug action Majority of drugs interact with target biomolecules: Usually a Protein ENZYMES ION CHANNELS TRANSPORTERS RECEPTORS

149
Enzymes – drug targets All Biological reactions are carried out under catalytic influence of enzymes Drugs – increases/decreases enzyme mediated reactions In physiological system Enzyme stimulation is less common by drugs – common by endogenous substrates Enzyme inhibition – common mode of drug action

150
Ion Channnels take part in transmembrane signaling and regulates ionic composition Drugs also target these channels: Ligand gated channels, G-protein operated channels, Direct action on channels

151
Transporters are translocated across membrane binding to specific transporters (carriers) – ,Pump the metabolites/ions In the direction of concentration gradient or against it
 – ,Pump the metabolites/ions In the direction of concentration gradient or against it.")
152
Receptors Drugs usually do not bind directly with enzymes, channels, transporters or structural proteins, but act through specific macromolecules- RECEPTORS Definition: It is defined as a macromolecule or binding site located on cell surface or inside the effector cell that serves to recognize the signal molecule/drug and initiate the response to it, but itself has no other function, e.g. G-protein coupled receptor

153
G-Protein coupled receptor
Membrane bound receptors which are bound to effector system through G-proteins. These are hetero trimeric molecules having 3 subunits α,β and ϒ. Based on α-sub unit they are further classified into 3 main varieties Gs, Gi and Gq

154
Subtypes of G-proteins - Targets (Second messenger systems)
Ion chanels: Na+ / H+ exchange Enzyms: Gi Inhib. Adenylyl cyclase Gs Stimul. Adenylyl cyclase Gq Stimul. Phospholipase C One ligand can bind to more than one type of G-proteins coupled receptors second messenger pathways

155
ENZYME LINKED RECEPTORS
Receptors intracellular domain is either protein kinase or guanyl cyclase Ex: Insulin, EGF, NGF Tyrosine Kinase binding receptors have no intrinsic catalytic domain but agonist induced dimerization affinity for cytosolic tyrosine kinase protein Ex:cytokines,growth hormone

156
TRANSCRIPTION RECEPTORS
Receptors regulating gene expression Intracellular- cytoplasmic or nuclear Ex:All steroid hormones,thyroxine, Vit A

157
FUNCTIONS OF RECEPTORS
To propogate signals from outside to inside To amplify the signal To integrate various extracellular and intracellular regulatory signals To adapt to long term changes in maintaining homeostasis

158
Drug+receptor drug-receptor complex effect
Lock and key theory

159
Affinity: Ability of a substrate to bind with receptor
Intrinsic activity (IA): Capacity to induce functional change in the receptor
: Capacity to induce functional change in the receptor.")
160
Agonist: An agent which activates a receptor to produce an effect similar to a that of the physiological signal molecule, e.g. Muscarine and Nicotine) response. Antagonist: an agent which prevents the action of an agonist on a receptor or the subsequent response, but does not have an effect of its own, e.g. atropine and muscarine no response.

163
Partial agonist: An agent which activates a receptor to produce submaximal effect but antagonizes the action of a full agonist, e.g. pentazocine Partial Inverse agonist: an agent which activates receptors to produce an effect in the opposite direction to that of the agonist, e.g. DMCM on bzp receptors opposite response Ligand: any molecule which attaches selectively to particular receptors or sites (only binding or affinity) If explained Agonist: Affinity+ IA Antagonist: Affinity+ IA (0) Partial agonist: Affinity + IA (0 to 1) Inverse agonist: Affinity + IA (0 to -1)
 If explained. Agonist: Affinity+ IA. Antagonist: Affinity+ IA (0) Partial agonist: Affinity + IA (0 to 1) Inverse agonist: Affinity + IA (0 to -1)")
165
Receptor regulation Desenstization or down-regulation
Supersenstivity or up-regulation

166
Enhancement of drug effects
A) Additive: 1+1= 2 B) Synergism: 1+1= 4 C) Potentiation: 1+0= 2
 Additive: 1+1= 2. B) Synergism: 1+1= 4. C) Potentiation: 1+0= 2.")
167
Adverse effects of drugs
Allergy: antigen-antibody………unpredictable Idiosyncrasy: genetic abnormality…….. Unpredictable Side effects: unavoidable, undesirable, normal actions by therapeutic doses. Over-dose: high dose of drugs Supersenstivity: exaggerated response to normal dose due to upregulation of receptors. Dependance: habituation and addiction.

168
Desensitization and Tachyphylaxis
When it is developing in the course of few minutes. Tolerance To describe a more gradual loss of drug-induced clinical effects that develops in the course of days or weeks. Refractoriness Used to indicate the loss of therapeutic response. Drug resistance Describes the loss of the effect of antitumour and antimicrobial drugs

169
MCQs G protein-coupled receptors that activate an inhibitory Gα subunit alter the activity of adenylyl cyclase to (A) increase the coupling of receptor to G protein (B) block the ligand from binding (C) initiate the conversion of GTP to GDP (D) generate intracellular inositol triphosphate (E) decrease the production of cAMP
 increase the coupling of receptor to G protein. (B) block the ligand from binding. (C) initiate the conversion of GTP to GDP. (D) generate intracellular inositol triphosphate. (E) decrease the production of cAMP.")
170
MCQs The law of mass action explains the relationship between (A) dose of drug and physiologic response (B) the concentration of drug and the association or dissociation of drug-receptor complex (C) receptors and the rate of signal transduction (D) an enzyme and ligands that inhibit the enzyme (E) graded and quantal dose-response curves
 the concentration of drug and the association or dissociation of drug-receptor complex. (C) receptors and the rate of signal transduction. (D) an enzyme and ligands that inhibit the enzyme. (E) graded and quantal dose-response curves.")
171
Apparent Volume of Distribution
Vd = Amount of drug in the body Plasma drug concentration VD = Dose/Plasma Concentration It is hypothetical volume of fluid in which the drug is disseminated. Units: L and L/Kg We consider the volume of fluid in the body = 60% of BW 60 X 70/100 = 42 L

172
Drug Distribution Water Body Compartments
Drugs may distribute into Plasma (Vascular) Compartment: Too large mol wt Extensive plasma protein binding Heparin is an example Extracellular Fluid Low mol wt drugs able to move via endothelial slits to interstitial water Hydrophilic drugs cannot cross cell membrane to the intracellular water Total Body Water; Low mol wt hydrophobic drugs distribute from interstitial water to intracellular Plasma (4 litres) Interstitial Fluid (11 litres) Intracellular Fluid (28 litres)
 Compartment: Too large mol wt. Extensive plasma protein binding. Heparin is an example. Extracellular Fluid. Low mol wt drugs able to move via endothelial slits to interstitial water. Hydrophilic drugs cannot cross cell membrane to the intracellular water. Total Body Water; Low mol wt hydrophobic drugs distribute from interstitial water to intracellular. Plasma. (4 litres) Interstitial Fluid. (11 litres) Intracellular Fluid. (28 litres)")
173
Distributed in plasma & Distributed in three comp.
Compartment Extracellular Compartment Intracellular Compartment Drug has large Mol. Wt. OR Bind extensively to pp Vd = 4L 6% of BW e.g. Heparin Drug has low Mol. Wt. Hydrophilic Distributed in plasma & Interstitial fluid Vd = 14L 21% of BW e.g. Aminoglycosides Drug has low Mol. Wt. Hydrophobic Distributed in three comp. Accumulated in fat Pass BBB Vd= 42L 60% of BW e.g. Ethanol

174
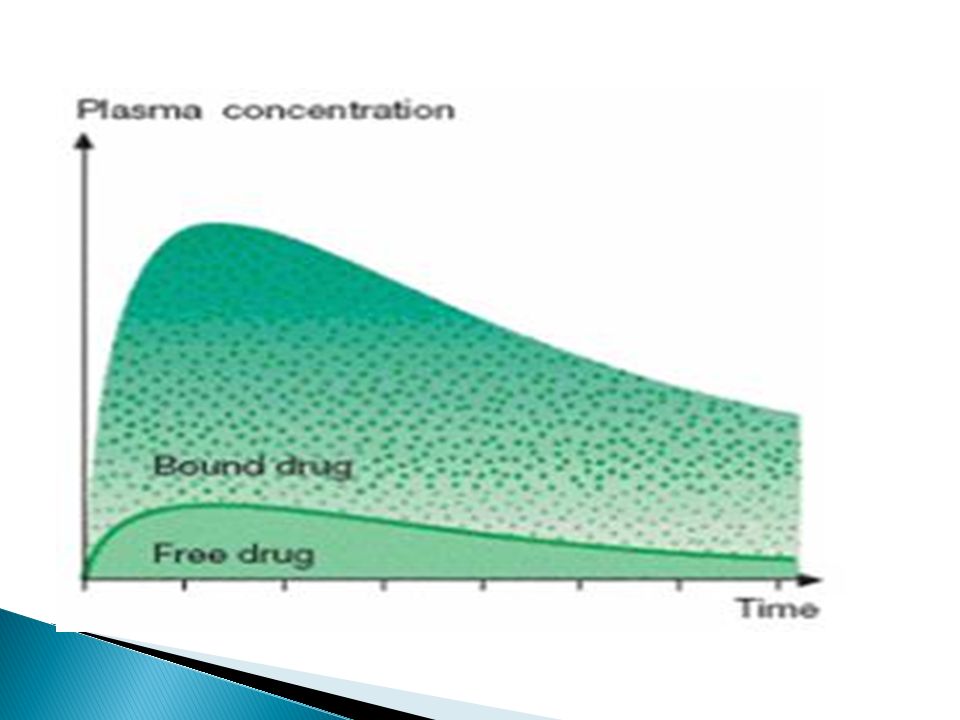
Plasma protein binding
Many drugs bind reversibly to plasma proteins especially albumin D + Albumin↔ D-Albumin (Inactive) + Free D Only free drug can distribute, binds to receptors, metabolized and excreted.
 + Free D. Only free drug can distribute, binds to receptors, metabolized and excreted.")
176
Clinical Significance of Albumin Biding
Class I: dose < available albumin binding sites (most drugs) Class II: dose > albumin binding sites (e.g., sulfonamide) Drugs of class II displace Class I drug molecules from binding sites→ more therapeutic/toxic effect In some disease states → change of plasma protein binding In uremic patients, plasma protein binding to acidic drugs is reduced Plasma protein binding prolongs duration Sulfonamide 176 Displacement of Class-I Drug
 Class II: dose > albumin binding sites (e.g., sulfonamide) Drugs of class II displace Class I drug molecules from binding sites→ more therapeutic/toxic effect. In some disease states → change of plasma protein binding. In uremic patients, plasma protein binding to acidic drugs is reduced. Plasma protein binding prolongs duration. Sulfonamide Displacement of Class-I Drug.")
177
Alter plasma binding of drugs
1000 molecules 900 999 % bound 1 molecules free 100 100-fold increase in free pharmacologically active concentration at site of action. Effective TOXIC

178
Capillary permeability
Endothelial cells of capillaries in tissues other than brain have wide slit junctions allowing easy movement of drugs Brain capillaries have no slits between endothelial cells, i.e tight junction or blood brain barrier Only carrier-mediated transport or highly lipophilic drugs enter CNS Ionised or hydrophilic drugs can’t get into the brain Liver capillary Slit junctions Brain capillary Glial cell Tight junctions Endothelial cells

179
Barriers to Drug Distribution
Blood-Brain barrier: Inflammation during meningitis or encephalitis may increase permeability into the BBB of ionised & lipid-insol drugs Placental Barrier: Drugs that cross this barrier reaches fetal circulation Placental barrier is similar to BBB where only lipophilic drugs can cross placental barrier

180
Metabolism It is enzyme catalyzed conversion of drugs to their metabolites. Process by which the drug is altered and broken down into smaller substances (metabolites) that are usually inactive. Lipid-soluble drugs become more water soluble, so they may be more readily excreted.
 that are usually inactive. Lipid-soluble drugs become more water soluble, so they may be more readily excreted.")
181
Most of drug biotransformation takes place in the liver, but drug metabolizing enzymes are found in many other tissues, including the gut, kidneys, brain, lungs and skin. Metabolism aims to detoxify the substance but may activate some drugs (pro-drugs).
.")
182
Reactions of Drug Metabolism
Phase I Phase II Conversion of Lipophyllic molecules Into more polar molecules by oxidation, reduction and hydrolysis reactions Conjugation with certain substrate ↑↓or unchanged Pharmacological Activity Inactive compounds

183
Phase I Biotransformation
Oxidative reactions: Catalyzed mainly by family of enzymes; microsomal cytochrome P450 (CYP) monoxygenase system. Drug + O2 + NADPH + H+ → Drugmodified + H2O + NADP+ Many CYP isoenzymes have been identified, each one responsible for metabolism of specific drugs. At least there are 3 CYP families and each one has subfamilies e.g. CYP3A. Many drugs alter drug metabolism by inhibiting (e.g. cimetidine) or inducing CYP enzymes (e.g. phenobarbital & rifampin). Pharmacogenomics
 monoxygenase system. Drug + O2 + NADPH + H+ → Drugmodified + H2O + NADP+ Many CYP isoenzymes have been identified, each one responsible for metabolism of specific drugs. At least there are 3 CYP families and each one has subfamilies e.g. CYP3A. Many drugs alter drug metabolism by inhibiting (e.g. cimetidine) or inducing CYP enzymes (e.g. phenobarbital & rifampin). Pharmacogenomics.")
184
Phase I Biotransformation (cont.)
Oxidative reactions: A few drugs are oxidised by cytoplasmic enzymes. Ethanol is oxidized by alcohol dehydrogenase Caffeine and theophylline are metabolized by xanthine oxidase Monoamine oxidase Hydrolytic reactions: Esters and amides are hydrolyzed by: Cholineesterase Reductive reactions: It is less common. Hepatic nitro reductase (chloramphenicol) Glutathione-organic nitrate reductase (NTG)
 Glutathione-organic nitrate reductase (NTG)")
185
Phase II Biotransformation
Drug molecules undergo conjugation reactions with an endogenous substrate such as acetate, glucuronate, sulfate or glycine to form water-soluble metabolites. Except for microsomal glucuronosyltransferase, these enzyems are located in cytoplasm. Most conjugated drug metabolites are pharmacologically inactive. Glucuronide formation: The most common using a glucuronate molecule. Acetylation by N-acetyltransferase that utilizes acetyl-Co-A as acetate donar. Sulfation by sulfotransferase. Sulfation of minoxidil and triamterene are active drugs.

186
Drug Excretion Excretion is the removal of drug from body fluids and occurs primarily in the urine. Other routes of excretion from the body include in bile, sweat, saliva, tears, feces, breast, milk and exhaled air.

Similar presentations

















 Absorption Distribution Metabolism Excretion Toxicology.>")
 - but the body is not homogeneous. Factors.>")















 The measure of the apparent space in the body available to contain.>")
