Download presentation
Presentation is loading. Please wait.

1
NGSS: HS-PS3 California State Standards Thermodynamics 7 Chapter 16
Energy NGSS: HS-PS3 California State Standards Thermodynamics 7 Chapter 16

2
Thermochemistry is the study of the transfers of energy as heat that accompany chemical reactions and physical changes.

3
Temperature is a measure of the average kinetic energy of the particles in a sample of matter.

4
What is a Thermometer? A thermometer is an instrument that measures the temperature of a system in a quantitative way. A substance having a property that changes in a regular way with its temperature is most useful; the most direct 'regular' way is a linear one: t(x) = ax + b, where t is the temperature of the substance and changes as the property x of the substance changes. The constants a and b depend on the substance used and may be evaluated by specifying two temperature points on the scale, such as 32° for the freezing point of water and 212° for its boiling point. For example, the element mercury is liquid in the temperature range of -38.9° C to 356.7° C. As a liquid, mercury expands as it gets warmer; its expansion rate is linear and can be accurately calibrated.
 = ax + b, where t is the temperature of the substance and changes as the property x of the substance changes. The constants a and b depend on the substance used and may be evaluated by specifying two temperature points on the scale, such as 32° for the freezing point of water and 212° for its boiling point. For example, the element mercury is liquid in the temperature range of -38.9° C to 356.7° C. As a liquid, mercury expands as it gets warmer; its expansion rate is linear and can be accurately calibrated.")
5
The mercury-in-glass thermometer illustrated in the above figure contains a bulb filled with mercury that is allowed to expand into a capillary. Its rate of expansion is calibrated on the glass scale

6
The Development of Thermometers and Temperature Scales
One of the first attempts to make a standard temperature scale occurred about AD 170, when Galen, in his medical writings, proposed a standard "neutral" temperature made up of equal quantities of boiling water and ice; on either side of this temperature were four degrees of heat and four degrees of cold, respectively. Claude Galien. Lithograph by Pierre Roche Vigneron. (Paris: Lith de Gregoire et Deneux, ca. 1865)
")
7
The earliest devices used to measure temperature were called
The earliest devices used to measure temperature were called thermoscopes. Glass bulb with a long tube extending downward into a container of colored water, (Galileo in 1610 is supposed to have used wine). Some of the air in the bulb was expelled before placing it in the liquid, causing the liquid to rise into the tube. As the remaining air in the bulb was heated or cooled, the level of the liquid in the tube would vary reflecting the change in the air temperature. An engraved scale on the tube allowed for a quantitative measure of the fluctuations. The air in the bulb is referred to as the thermometric medium, i.e. the medium whose property changes with temperature. Portrait of Galileo Galilei by Giusto Sustermans
. Some of the air in the bulb was expelled before placing it in the liquid, causing the liquid to rise into the tube. As the remaining air in the bulb was heated or cooled, the level of the liquid in the tube would vary reflecting the change in the air temperature. An engraved scale on the tube allowed for a quantitative measure of the fluctuations. The air in the bulb is referred to as the thermometric medium, i.e. the medium whose property changes with temperature. Portrait of Galileo Galilei. by. Giusto Sustermans.")
8
Robert Hook, Curator of the Royal Society, in 1664 used a red dye in the alcohol. His scale, for which every degree represented an equal increment of volume equivalent to about 1/500 part of the volume of the thermometer liquid, needed only one fixed point. He selected the freezing point of water. By scaling it in this way, Hook showed that a standard scale could be established for thermometers of a variety of sizes. Hook's original thermometer became known as the standard of Gresham College and was used by the Royal Society until (The first intelligible meteorological records used this scale).
..")
9
In 1702, the astronomer Ole Roemer of Copenhagen based his scale upon two fixed points: snow (or crushed ice) and the boiling point of water, and he recorded the daily temperatures at Copenhagen in with this thermometer.

10
It was in 1724 that Gabriel Fahrenheit, an instrument maker of Däanzig and Amsterdam, used mercury as the thermometric liquid. Fahrenheit measured the b.p. of water to be 212. He adjusted the f.p. to 32 so that the interval between the boiling and freezing points of water could be 180. Temperatures measured on this scale are designated as degrees Fahrenheit (°F).
.")
11
In 1745, Carolus Linnaeus (Swedish), f. p. = 0; b
In 1745, Carolus Linnaeus (Swedish), f.p. = 0; b.p = 100 (for H2O), a centigrade (one hundred steps) scale. Anders Celsius ( ) used the reverse scale in which f.p. = 100 and b.p. = 0, still with 100 degrees between the two defining points.
, f.p. = 0; b.p = 100 (for H2O), a centigrade (one hundred steps) scale. Anders Celsius ( ) used the reverse scale in which f.p. = 100 and b.p. = 0, still with 100 degrees between the two defining points.")
12
In 1948 use of the Centigrade scale was dropped in favor of a new scale using degrees Celsius (°C).
On the Celsius scale the boiling point of water at standard atmospheric pressure is °C in contrast to the 100 degrees defined by the Centigrade scale. To convert from Celsius to Fahrenheit: ° F = (1.8 x °C) + 32
")
13
Another temperature scale is the Kelvin temperature scale (absolute temperature scale), after Lord Kelvin William Thompson ( ). K = °C + 273

14
Heat and Thermodynamics
Prior to the 19th century, it was believed that the sense of how hot or cold an object felt was determined by how much "heat" it contained. Heat was envisioned as a liquid that flowed from a hotter to a colder object In 1847 J. P. Joule published a definitive paper that conclusively showed that heat was a form of energy, which could be transformed from one form into another.

15
First law of thermodynamics
When heat is transformed into any other form of energy, or when other forms of energy are transformed into heat, the total amount of energy (heat plus other forms) in the system is constant. This is also referred to as the law of conservation of energy.
 in the system is constant. This is also referred to as the law of conservation of energy.")
16
[N = Newton(unit of force; F = ma)]
Energy is the ability to do work or produce heat; kinetic or potential energy. Joule = N x m [N = Newton(unit of force; F = ma)] = kg x m2 s2 The amount of energy transferred as heat is usually measured in joules Heat can be thought of as the amount of energy transferred between samples of matter because of a difference in their temperatures.
![[N = Newton(unit of force; F = ma)]](http://slideplayer.com/slide/7016034/24/images/16/%5BN+%3D+Newton%EF%83%A0%28unit+of+force%3B+F+%3D+ma%29%5D.jpg "Energy is the ability to do work or produce heat; kinetic or potential energy. Joule = N x m. [N = Newton(unit of force; F = ma)] = kg x m2. s2. The amount of energy transferred as heat is usually measured in joules Heat can be thought of as the amount of energy transferred between samples of matter because of a difference in their temperatures.")
17
Calorimetry. is a measurement of the amount of heat released or absorbed during chemical reactions. A calorimeter is used to measure this amount of heat

18
Specific heat capacity
The amount of energy required to raise the temperature of one gram of substance by one degree Celsius (1 ºC) Values can be expressed in J/(g ·ºC) The equation used to measure the amount of energy gained or lost with a change in temperature is q = m · cp · ∆T
 Values can be. expressed in J/(g ·ºC) The equation used to measure the amount of energy gained or lost with a change in temperature is. q = m · cp · ∆T.")
19
thermochemical equation
2H2(g) + O2 (g) 2H2O (g) kJ or 2H2(g) + O2 (g) 2H2O (g) ∆H = kJ Exothermic reaction
 + O2 (g) 2H2O (g) kJ. or. 2H2(g) + O2 (g) 2H2O (g) ∆H = kJ. Exothermic reaction.")
20
thermochemical equation
2H2O (g) kJ 2H2(g) + O2 (g) or 2H2O (g) 2H2(g) + O2 (g) ∆H = kJ Endothermic reaction
 kJ 2H2(g) + O2 (g) or 2H2O (g) 2H2(g) + O2 (g) ∆H = kJ Endothermic reaction")
21
Reaction pathways

22
Molar heat of formation
the energy released or absorbed as heat when one mole of a compound is formed from its elements. The standard heat of formation is when it is measured with the substances in their standard states. ∆H ºf

23
Heat of combustion. The energy released as heat by the complete combustion of one mole of a substance. ∆H ºc The Heat of combustion is defined in terms of one mole of reactant, whereas the heat of formation is defined in terms of one mole of product

24
∆H ºrxn = ∑∆H ºf (products) – ∑∆H ºf (reactants)
 – ∑∆H ºf (reactants)")
25
Sample problem If the temperature of 34.4 g of ethanol increases from 25.0 ºC to 78.8 ºC, how much heat (in Joules) has been absorbed by the ethanol? How many Calories is this equal to?

26
q = m · cp · ∆T (34.4 g)(2.44 J/g ºC )(53.8 ºC) = J J x 1 cal x 1 kcal = J 1000 cal Calories
(2.44 J/g ºC )(53.8 ºC) = J J x 1 cal x 1 kcal = J 1000 cal Calories")
27
Sample problem #2 #18 from text page 553

28
Hess’s Law The overall enthalpy change in a reaction is
equal to the sum of enthalpy changes for the individual steps in the process Standard enthalpies of formation provide useful data for calculating the enthalpies of reactions under standard conditions (∆H ºrxn) using Hess’s law.
 using Hess’s law.")
29
If a reaction is reversed, the sign of ∆H is also reversed.
The rules for combining thermochemical equations are as follows: If a reaction is reversed, the sign of ∆H is also reversed. Multiply the coefficients of the known equations so that when added together they give the desired thermochemical equation.

30
∆H ºrxn = ∑∆H ºf (products) - ∑∆H ºf (reactants)
Example: Calculate the heat of formation of methane gas, CH4, from its elements at K (25ºC) We need to use the combustion reactions of the elements, carbon and hydrogen, and of methane. C(s) + O2(g) CO2(g) ∆Hºc = kJ/mol H2(g) + 1/2O2(g) H2O (l) ∆Hºc = kJ/mol CH4(g) + 2O2(g) CO2(g) + 2H2O(l) ∆Hºc = kJ/mol
 We need to use the combustion reactions of the elements, carbon and hydrogen, and of methane. C(s) + O2(g) CO2(g) ∆Hºc = kJ/mol. H2(g) + 1/2O2(g) H2O (l) ∆Hºc = kJ/mol. CH4(g) + 2O2(g) CO2(g) + 2H2O(l) ∆Hºc = kJ/mol.")
31
C(s) + 2H2(g) CH4(g) ∆H ºf = -74.3 kJ/mol
C(s) + O2(g) CO2(g) ∆H ºc = kJ/mol 2[H2(g) + 1/2O2(g) H2O (l) ∆H ºc = kJ/mol] CO2(g) + 2H2O(l) CH4(g) + 2O2(g)∆H ºc = kJ/mol C(s) + 2H2(g) CH4(g) ∆H ºf = kJ/mol
 + O2(g) CO2(g) ∆H ºc = kJ/mol. 2[H2(g) + 1/2O2(g) H2O (l) ∆H ºc = kJ/mol] CO2(g) + 2H2O(l) CH4(g) + 2O2(g)∆H ºc = kJ/mol. C(s) + 2H2(g) CH4(g) ∆H ºf = kJ/mol.")
32
Molar heat of vaporization: the amount of heat energy needed to vaporize one mole of a liquid at its boiling point. Molar heat of fusion: the amount of heat energy needed to melt one mole of solid at its melting point.

33
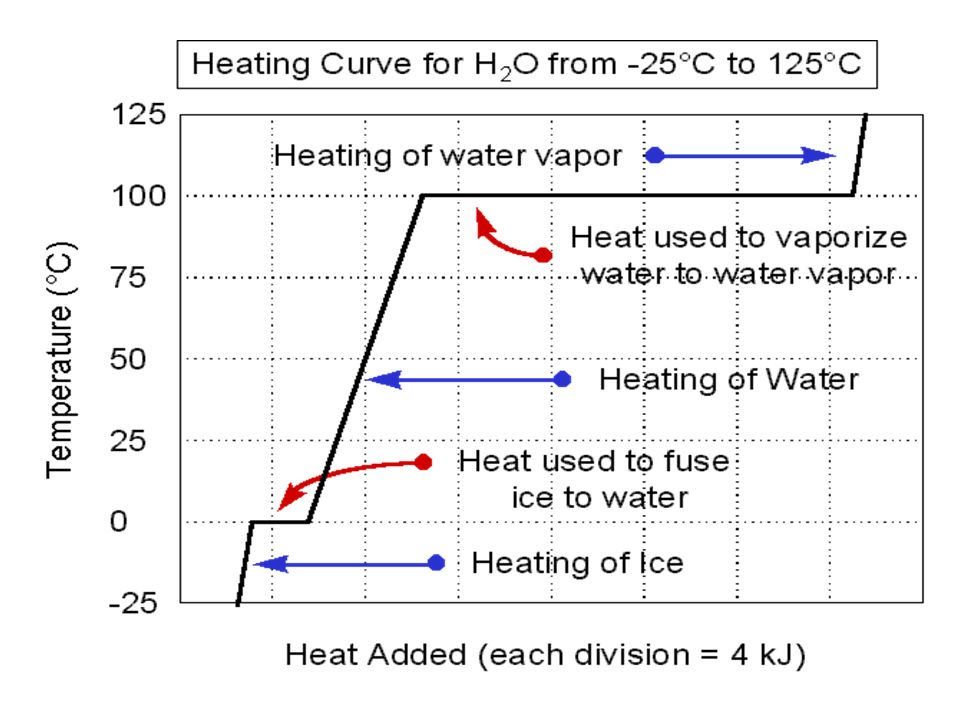
Heating ice, water and water vapor
The amount of heat needed to change the temperature of a substance is given by the specific heat or molar heat capacity Specific heat of ice = 2.06 J/g K Specific heat of water = J/g K Specific heat of water vapor = 1.87 J/g K In the regions of the curve where we are undergoing a phase transition, the heat energy input is not raising the temperature of the sample; rather it is being used to disrupt the intermolecular forces ∆Hfus = 6.01 kJ/mol ∆Hvap = 40.7 kJ/mol

35
Entropy Measure of the degree of randomness of the particles in a system (kJ/ K) Entropy of a pure crystalline solid is zero at O K.

36
ΔSsystem = Sproducts – Sreactants
ΔS+ increase in entropy ΔS- decrease in entropy Increase in entropy – forming a solution as you are increasing the randomness of the system; also mixing gases, and dissolving a liquid in a liquid, and dissolving a solid in a liquid

37
2nd law of thermodynamics
(law of disorder): spontaneous processes always proceed in such a way that the entropy of the universe increases.
: spontaneous processes always proceed in such a way that the entropy of the universe increases.")
38
ΔSuniverse = ΔSsystem + ΔSsurroundings
The following is true for any spontaneous process ΔS universe > 0 Universe = system + surroundings ΔSuniverse = ΔSsystem + ΔSsurroundings If a rxn is exothermic, ΔH is negative (which increases the temp. of the surroundings) so Δssurroundings is positive. The entropy of the system increases as well, so Δssystem is also positive.
 so Δssurroundings is positive. The entropy of the system increases as well, so Δssystem is also positive.")
39
Free energy Energy available to do work (1878 –Willard Gibbs - Yale) – useful energy ΔGsystem = ΔHsystem – TΔSsystem If the reaction occurs under standard conditions, 298 K and 1 atm pressure) ΔGºsystem = ΔHºsystem – TΔSºsystem
 – useful energy ΔGsystem = ΔHsystem – TΔSsystem If the reaction occurs under standard conditions, 298 K and 1 atm pressure) ΔGºsystem = ΔHºsystem – TΔSºsystem")
40
ΔGsystem and reaction spontaneity
-ΔGsystem = spontaneous +ΔGsystem = nonspontaneous ΔGsystem and reaction spontaneity Type of reaction or process ΔGsystem ΔSuniverse Spontaneous negative positive Nonspontaneous positive negative

41
ΔGsystem = ΔHsystem – TΔSsystem
How ΔHsystem and ΔSsystem affect reaction spontaneity -ΔHsystem +ΔHsystem +ΔSsystem Always spontaneous Spontaneity depends upon temp. -ΔSsystem Never spontaneous

42
Energy Websites 13 min THIS IS THE ONE TO WATCH!!! The rest are also helpful! Chemistry: CrashCourse – a whole bunch of excellent videos Bozeman science entropy min 2min

43
Reaction Kinetics Standard 8
Chapter 17 Reaction Kinetics Standard 8

44
Isaacs TEACH collision theory 5:12 potential energy diagrams 6:54 rate of reaction and rate laws 11:14 (continue watching through the accelerated class section!) factors that affect reaction rate 10:38 (continue watching through the accelerated class section!) reaction mechanisms (and rate limiting step) 4:31 Crash Course Chemistry: Chemistry's Demolition Derby - 9:57 This also introduces/reviews some equilibrium concepts!
 v=S1sWvCOTUl8 – factors that affect reaction rate 10:38 (continue watching through the accelerated class section!) v=eEFJpmFk-9A – reaction mechanisms (and rate limiting step) 4:31. Crash Course Chemistry: v=7qOFtL3VEBc Kinetics: Chemistry s Demolition Derby - 9:57 This also introduces/reviews some equilibrium concepts!")
45
Bozeman Science Rate Law 8:43 (Don’t worry about finding slope of curves – beyond what you need to know) – watch this one before the one on the Rate Constant rate of Reactions 6:24 (you do NOT need to know anything about Beers Law!) of Reaction 8:02 Review of Chapter 16 material (calorimetry, Hess’s law) Rate Constant 6:52 (a bit more in depth starting about halfway through the video) If you’re going to skip a video, this one would be it!
 – watch this one before the one on the Rate Constant. v=6mAqX31RRJU The rate of Reactions 6:24 (you do NOT need to know anything about Beers Law!) v=qD7PDOhqbpM Enthalpy of Reaction 8:02 Review of Chapter 16 material (calorimetry, Hess’s law) v=eOSRn0jPbTk The Rate Constant 6:52 (a bit more in depth starting about halfway through the video) If you’re going to skip a video, this one would be it!")
46
Brightstorm videos: reaction rates factors 3:04 Reaction rate problems 9:38 Excellent! Look at primarily the second problem (which is quite involved)
")
47
Average reaction rate = -∆ quantity ∆t
Reaction Rates change in concentration of reactants per unit time as a reaction proceeds. Average reaction rate = -∆ quantity ∆t Chemical kinetics is the area of chemistry that deals with reaction rates and reaction mechanisms.

48
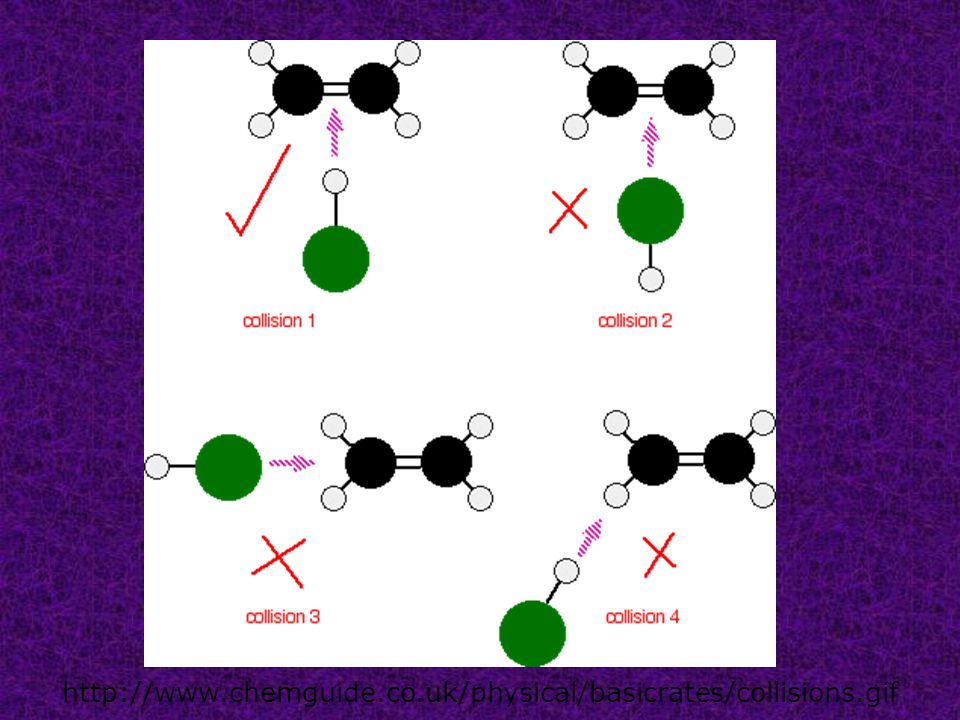
Collision theory states that in order for reactions to occur between substances their particles must collide with sufficient energy and with a favorable orientation.

50
Exothermic Reaction pathway

51
Endothermic reaction pathway

52
Activation energy : minimum energy required to transform the reactants into an activated complex.

53
Activated complex transitional structure that results from an effective collision and that persists while old bonds are breaking and new bonds are forming.

54
Factors affecting reaction rate:
For homogeneous reactions Nature of reactants: the types of reactants involved (i.e. ions vs. molecular substances; gases vs. liquids or solids)
")
55
Concentration: the higher the concentration of one or more of the reactants results the faster the reaction rate.

56
Temperature An increase in temperature increases the number of particles that have enough energy to form the activated complex. In general, for every 10 K (10˚C) rise in temperature, there is a doubling of the rate.
 rise in temperature, there is a doubling of the rate.")
57
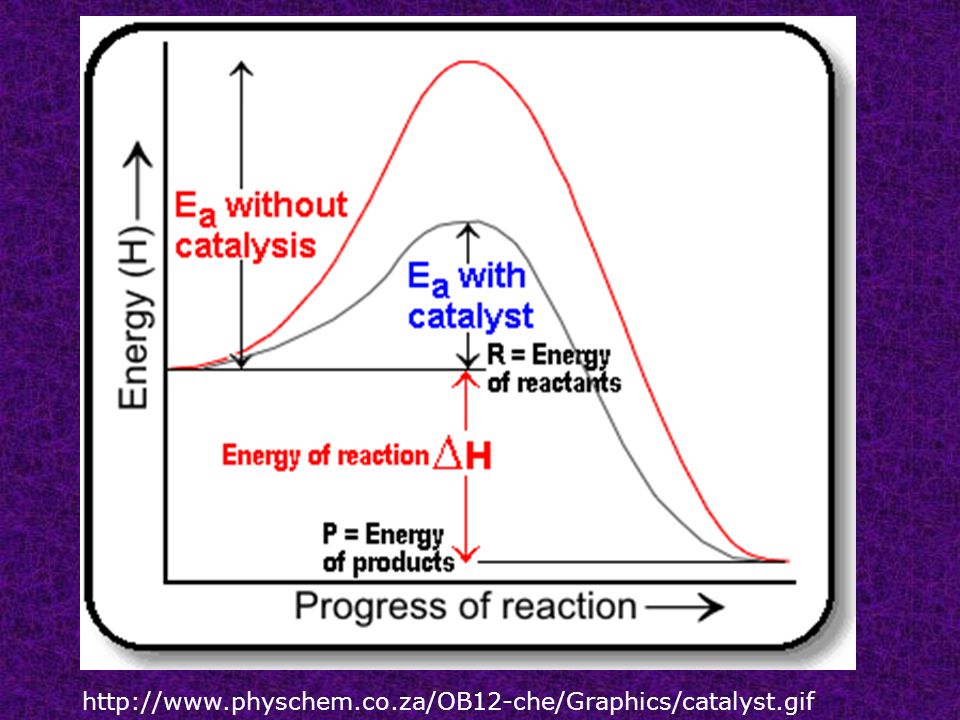
Catalysts substances that change the rate of the reaction without being consumed. Homogeneous catalysts are of the same phase as the reactants while heterogeneous catalysts are of a different phase as the reactants.

59
Enzyme animation:

60
For heterogeneous reactions:
surface area is also important. (i.e. wood chips vs. a log)
")
61
Reaction Mechanisms step-by-step sequence of reactions by which the overall chemical change occurs

62
Rate-determining step.
The slowest step in multiple step chemical reaction. This can be determined experimentally or you would be given this information.

63
Intermediates Species that appear in some steps but not in the net ionic equation.

64
Two possible mechanisms
I2 2I- 2I- + H2 2HI I2 + H2 2HI I2 2I- I- + H2 H2I H2I + I- 2HI I2 + H2 2HI I- and H2I are both intermediates

65
Reaction Rate Laws A rate law is an equation that relates reaction rate and concentrations of reactants. It is specific for reactions at specific temperatures.

66
Example: R = k[A][B] Single step reaction.
Suppose A + B 2C. One particle of A and B is involved in each collision. So, doubling the concentration of either A or B will double the collision frequency, thus doubling the reaction rate. Therefore the rate law is R = k[A][B]
![Example: R = k[A][B] Single step reaction.](http://slideplayer.com/slide/7016034/24/images/66/Example%3A+R+%3D+k%5BA%5D%5BB%5D+Single+step+reaction..jpg "Suppose A + B 2C. One particle of A and B is involved in each collision. So, doubling the concentration of either A or B will double the collision frequency, thus doubling the reaction rate. Therefore the rate law is. R = k[A][B]")
67
Example continued R = k[C] 2
If the reaction is reversible, 2C A + B, then two molecules of C must decompose to form one each of A and B. Thus the rate law is R = k[C] 2
![Example continued R = k[C] 2](http://slideplayer.com/slide/7016034/24/images/67/Example+continued+R+%3D+k%5BC%5D+2.jpg "If the reaction is reversible, 2C A + B, then two molecules of C must decompose to form one each of A and B. Thus the rate law is. R = k[C] 2.")
68
Reaction order The exponents to which the concentration terms are raised (define how the rate is affected by the concentration of that reactant), and their sum is the overall reaction order. The value of these exponents is always determined experimentally.
, and their sum is the overall reaction order. The value of these exponents is always determined experimentally.")
69
zero-order changing the concentration of one reactant has no effect on the rate, then it is zero-order for that reactant

70
1st order rate depends on the concentration of a single reactant raised to the first power A products Rate = k[A]

71
2nd order rate depends on the reactant concentration raised to the second power or on the concentrations of two different reactants, each raised to the first power Rate = k[A]2

72
General form for rate law expression
Rate = k [A]m[B]n Determine values for m and n

73
Example problem The initial rate of a reaction A + B C was measured for several different starting concentrations of A and B, with the results given here:

74
Experiment number [A] (M) [B] (M) Initial rate (M/s) 1 0.100 4.0 x 10-5 2 0.200 3 16.0 x 10-5 Using these data, determine (a) the rate law for the reaction; (b) the magnitude of the rate constant; (c) the rate of the reaction when [A] = M and [B]= M.
![Experiment number [A] (M) [B] (M) Initial rate. (M/s) x x](http://slideplayer.com/slide/7016034/24/images/74/Experiment+number+%5BA%5D+%28M%29+%5BB%5D+%28M%29+Initial+rate.+%28M%2Fs%29+x+x.jpg "Using these data, determine (a) the rate law for the reaction; (b) the magnitude of the rate constant; (c) the rate of the reaction when. [A] = M and [B]= M.")
75
Solution part A Looking at experiment 1 and 2 keeping [A] constant, doubling [B]; no effect on rate. Order of B is 0 (n=0). Looking at experiments 1 and 3 keeping [B] constant, doubling [A]; rate increased by a factor of 4, i.e. [A]2 (m=2). rate = k[A]2
. Looking at experiments 1 and 3 keeping [B] constant, doubling [A]; rate increased by a factor of 4, i.e. [A]2 (m=2). rate = k[A]2.")
76
Solution part B [A]2 = 4.0 x 10-5 M/s (0.100 M)2 = 4.0 x 10-3 M-1s-1
k = rate [A]2 = 4.0 x 10-5 M/s (0.100 M)2 = 4.0 x 10-3 M-1s-1
![Solution part B [A]2 = 4.0 x 10-5 M/s (0.100 M)2 = 4.0 x 10-3 M-1s-1](http://slideplayer.com/slide/7016034/24/images/76/Solution+part+B+%5BA%5D2+%3D+4.0+x+10-5+M%2Fs+%280.100+M%292+%3D+4.0+x+10-3+M-1s-1.jpg "k = rate. [A]2. = 4.0 x 10-5 M/s. (0.100 M)2. = 4.0 x 10-3 M-1s-1.")
77
Solution part C Rate = k[A]2 (4.0 x 10-3 M-1s-1)(0.050 M)2 = 1.0 x 10-5 M/s
![Solution part C Rate = k[A]2 (4.0 x 10-3 M-1s-1)(0.050 M)2 = 1.0 x 10-5 M/s](http://slideplayer.com/slide/7016034/24/images/77/Solution+part+C+Rate+%3D+k%5BA%5D2+%284.0+x+10-3+M-1s-1%29%280.050+M%292+%3D+1.0+x+10-5+M%2Fs.jpg "Solution part C Rate = k[A]2 (4.0 x 10-3 M-1s-1)(0.050 M)2 = 1.0 x 10-5 M/s")
78
Isaacs TEACH collision theory 5:12 potential energy diagrams 6:54 rate of reaction and rate laws 11:14 (continue watching through the accelerated class section!) factors that affect reaction rate 10:38 (continue watching through the accelerated class section!) reaction mechanisms (and rate limiting step) 4:31
 v=S1sWvCOTUl8 – factors that affect reaction rate 10:38 (continue watching through the accelerated class section!) v=eEFJpmFk-9A – reaction mechanisms (and rate limiting step) 4:31.")
79
Crash Course Chemistry: https://www. youtube. com/watch
Crash Course Chemistry: Chemistry's Demolition Derby - 9:57 This also introduces/reviews some equilibrium concepts!

80
Bozeman Science Rate Law 8:43 (Don’t worry about finding slope of curves – beyond what you need to know) – watch this one before the one on the Rate Constant rate of Reactions 6:24 (you do NOT need to know anything about Beers Law!) of Reaction 8:02 Review of Chapter 16 material (calorimetry, Hess’s law) Rate Constant 6:52 (a bit more in depth starting about halfway through the video) If you’re going to skip a video, this one would be it!
 – watch this one before the one on the Rate Constant. v=6mAqX31RRJU The rate of Reactions 6:24 (you do NOT need to know anything about Beers Law!) v=qD7PDOhqbpM Enthalpy of Reaction 8:02 Review of Chapter 16 material (calorimetry, Hess’s law) v=eOSRn0jPbTk The Rate Constant 6:52 (a bit more in depth starting about halfway through the video) If you’re going to skip a video, this one would be it!")
81
Brightstorm videos: reaction rates factors 3:04 Reaction rate problems 9:38 Excellent! Look at primarily the second problem (which is quite involved)
")
Similar presentations















 exothermic/endothermic calorie/joule heat capacity/specific heat 17.2– Measuring and.>")



