Download presentation
Presentation is loading. Please wait.

1
Docking@GRID – A Web Portal for Massively Parallel Flexible Docking, using the ChemAxon toolkit. Dragos Horvath*, Kun Attila, Benjamin Parent*, Cyrielle Boutroue #, Even Gaël #, Alexandru Tantar #, Nouredine Melab #, Sylvaine Roy & El-Ghazali Talbi # * UMR 8576, CNRS – Univ. Lille 1, FR Chemistry Dept, Univ. Babes-Bolyai, Cluj, RO Chemistry Dept, Univ. Babes-Bolyai, Cluj, RO # LIFL CNRS/INRIA – Univ. Lille 1, FR DSV/iRTSV - CEA, Grenoble, FR DSV/iRTSV - CEA, Grenoble, FR

2
Outline… The goal: automated fully flexible docking on computer grids –GRID5000, http://www.grid5000.frhttp://www.grid5000.fr –Specific conformational sampling & docking software based on hybrid genetic algorithms –Upfront chemoinformatics tools to preprocess submitted ligands. –Upfront tools to define the active site and its key degrees of freedom (!) –Interface to start docking calculations & analyze results.
 –Interface to start docking calculations & analyze results..")
3
Genetic Algorithm-driven Conformational Sampling Tool Based on a Genetic Algorithm, coding conformers as "chromosomes" in which each locus stands for a torsional angle value. n … The In Silico Darwinian Evolution, leading to fitter and fitter (lower energy) conformers, was enhanced by –hybridization with various optimization heuristics –Fine-tuning of the parameters controlling the evolutionary strategy Customized CVFF force field, employing: a 10 Å cutoff (with a termination function) a smoothing procedure to avoid interatomic clashes a continuum solvent model Effective interatomic distance d 0 ij Smoothing distance d ij
 conformers, was enhanced by –hybridization with various optimization heuristics –Fine-tuning of the parameters controlling the evolutionary strategy Customized CVFF force field, employing: a 10 Å cutoff (with a termination function) a smoothing procedure to avoid interatomic clashes a continuum solvent model Effective interatomic distance d 0 ij Smoothing distance d ij.")
4
GRID 5000-based Planetary Model If (free node) DEPLOY Island Model - Executables - Molecule File - Constraint Files - Seeds List - Taboo List - Operational Pars -Stablest Chromosomes -Sampling Success Score Solution Merger & Clusterer Conformer & Cluster Database Panspermia policy center recent clusters: seeds old clusters: taboo Sampling Success vs. Operational Pars Stop: max. Mission Nr. no new clusters since N missions www.grid5000.fr Operational Pars Selector

5
Ab initio folding of Trp cage 1L2Y: native structure (reproducibly) found and ranked as most stable. Planetary model used max. 20 nodes for 4…5 daysAb initio folding of Trp cage 1L2Y: native structure (reproducibly) found and ranked as most stable. Planetary model used max. 20 nodes for 4…5 days Conformer # 1, RMS~1.8 Ǻ - good match to native structure
 found and ranked as most stable. Planetary model used max. 20 nodes for 4…5 days Conformer # 1, RMS~1.8 Ǻ - good match to native structure.")
6
Ab initio folding of Trp zipper 1LE1: native structure found and ranked as most stable. Planetary model used max. 20 nodes for 4…5 daysAb initio folding of Trp zipper 1LE1: native structure found and ranked as most stable. Planetary model used max. 20 nodes for 4…5 days Conformer # 1, RMS~0.8 Ǻ - perfect match to native structure

7
However, there is a high risk that almost well folded solutions, being declared taboo, block the access to the correct fold !!However, there is a high risk that almost well folded solutions, being declared taboo, block the access to the correct fold !! Conformer # 79, RMS~2.4 Ǻ - near-optimal fold closest to native structure Conformer # 1, RMS~3.8 Ǻ - is a poor match of the native structure

8
Outline… The goal: automated fully flexible docking on computer grids –GRID5000, http://www.grid5000.frhttp://www.grid5000.fr –Specific conformational sampling & docking software based on hybrid genetic algorithms –Upfront chemoinformatics tools to pre- process submitted ligands. –Upfront tools to define the active site and its key degrees of freedom (!) –Interface to start docking calculations & analyze results.
 –Interface to start docking calculations & analyze results..")
9
Ligand Preprocessor… Ligand File Upload Standardize Main Tautomer & Key µSpecies (occurrence > m%) All Tautomers & Major µSpecies All Tautomers & Key µSpecies Add Explicit H Force Field Typing (PMapper) JChem DataBase Cannonical SMILES Dockable Conformer Families Main Tautomer & Major µSpecies User Toggle Partial Charge Calculation Generate Conformer(s) If new… A selector of top N most likely tautomeric forms would be of outstanding help here – many among the enumenated tautomers are chemically meaningless! Potential problems with resonant structures in the ChargePlugin: try { ChgPlug.setTakeResonantStructure(true); chgMol=ChgPlug.setMolecule(currSpec,false,false); ChgPlug.run(); … } catch (Exception ResonantStructureFailed) { try { ChgPlug.setTakeResonantStructure(false); … } catch (Exception WhateverYouDoItBreaks) { … } Using PMapper to assign CVFF types to ligand atoms required SMARTS encoding of the CVFF templates corresponding to local neighborhoods defining each potential type Issues yet to be settled: use the Conformer Plugin to generate several hundreds of geometries Conformer diversity control ? How many degrees of freedom can be handled without significant risk of missing key minima ? Docking will use a different force field – how compatible are ConformerPlugin & CVFF energies? use the Conformer Plugin to generate a starting geometry, then use a ligand-specific GA-driven sampling engine to explore the phase space.
; chgMol=ChgPlug.setMolecule(currSpec,false,false); ChgPlug.run(); … } catch (Exception ResonantStructureFailed) { try { ChgPlug.setTakeResonantStructure(false); … } catch (Exception WhateverYouDoItBreaks) { … } Using PMapper to assign CVFF types to ligand atoms required SMARTS encoding of the CVFF templates corresponding to local neighborhoods defining each potential type Issues yet to be settled: use the Conformer Plugin to generate several hundreds of geometries Conformer diversity control . How many degrees of freedom can be handled without significant risk of missing key minima . Docking will use a different force field – how compatible are ConformerPlugin & CVFF energies. use the Conformer Plugin to generate a starting geometry, then use a ligand-specific GA-driven sampling engine to explore the phase space..")
11
Outline… The goal: automated fully flexible docking on computer grids –GRID5000, http://www.grid5000.frhttp://www.grid5000.fr –Specific conformational sampling & docking software based on hybrid genetic algorithms –Upfront chemoinformatics package to pre- process submitted ligands. –Upfront tools to define the active site and its key degrees of freedom (!) –Interface to start docking calculations & analyze results.
 –Interface to start docking calculations & analyze results..")
12
Active Site Definition… Ligand.. Fixed protein residues Fixed backbone, Mobile sidechains Flexible Loop: Backbone ( but not ) & sidechains This part of the backbone is a « frozen » part of the flexible loop: Rigid body rototranslations Formally « break » bond to unlock degrees of freedom in loop
 & sidechains This part of the backbone is a « frozen » part of the flexible loop: Rigid body rototranslations Formally « break » bond to unlock degrees of freedom in loop.")
13
Protein Preprocessing Tools… At this point, the user has to explicitly provide: –A BioSym.car protein file, with correct protonation states, partial charges and force field types for all protein atoms –A list of fixed atoms –A list of explicitly broken bonds to enable sampling ring and fixed end loop geometries –A list of active torsional degrees of freedom (otherwise, all potentially rotatable exocyclic single bonds will be considered) Will MarvinSpace evolve such as to allow for graphical input the above-mentioned information? Would the Charge Plugin, the MicroSpecies Plugin and PMapper work upon input of a.pdb file? JChem Database of defined active sites and their sampled unbound state geometries…

14
Outline… The goal: automated fully flexible docking on computer grids –GRID5000, http://www.grid5000.frhttp://www.grid5000.fr –Specific conformational sampling & docking software based on hybrid genetic algorithms –Upfront chemoinformatics tools to pre-process submitted ligands. –Upfront tools to define the active site and its key degrees of freedom (!) –Interface to start docking calculations & analyze results.
 –Interface to start docking calculations & analyze results..")
15
The Dock Manager In an ideal world, an academic user may add own molecule collections to the database, but should be allowed to try docking other peoples molecules as well… –Paranoia Manager: whos allowed to dock my compounds and use my active sites? –Make use of JChem facilities to search ligand database by cannonical structures, and return all the conformers of associated µSpecies/Tautomers. Chemoinformatic filters welcome, even based on the Holy Rule of Five! Methodological progress on the docking algorithms still required: –Is rigid docking of each of ~10 2 ligand conformers into each one of the ~10 4 active site geometries feasible? Would it be assimilable to flexible docking? –How to score: free energy based on docked vs. unbound ensembles? What about µSpecies & Tautomer penalties?

16
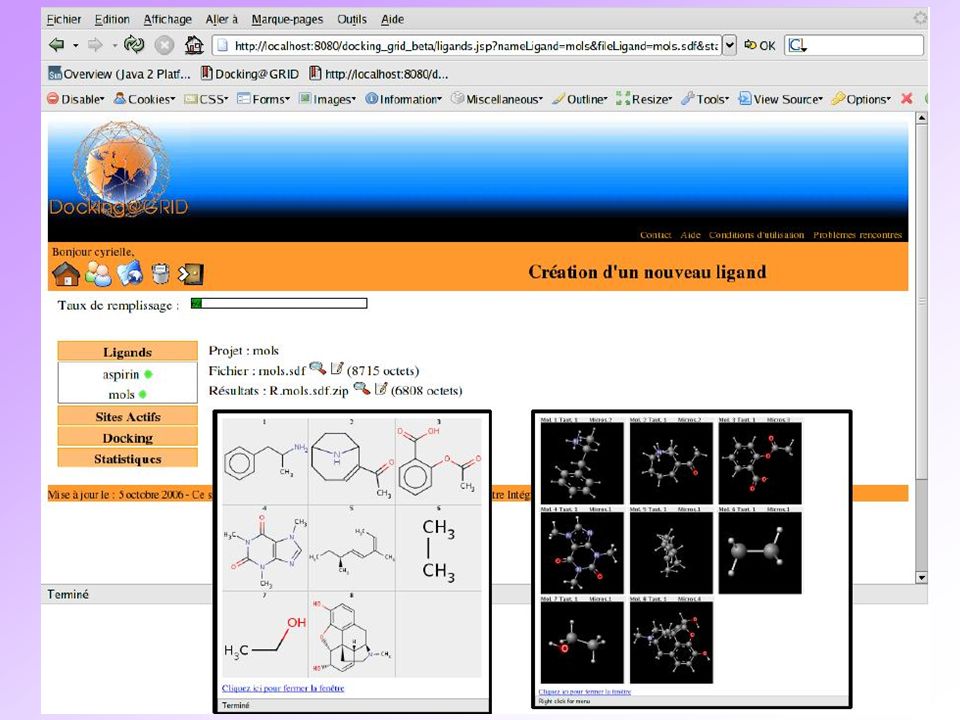
Docked Conformer Visualization

17
Conclusions & Perspectives This is a long-term ANR-funded public research project: http://dockinggrid.gforge.inria.fr/http://dockinggrid.gforge.inria.fr/ The primary goal is developing efficient GRID-based conformational sampling & docking methodologies –http://paradiseo.gforge.inria.fr/ to provide the core routines for parallel evolutionary computinghttp://paradiseo.gforge.inria.fr/ However, chemically meaningful ligand and active site management is as important as the docking step! –ChemAxon tools for ligand standardizing, protonation, charge & force field management, 3D-buildup, storage & retrieval, visualizing,…, are perfectly suited! –Progress needed on macromolecule & active site management. T T H A N K S H T H A N K S A T H A N K S N T H A N K S K T H A N K S S

Similar presentations
 – ANR meeting – META08 Hammamet 1/18 validation ANR meeting - 28/10/2008 CEA Grenoble - DSV/iRTSV/CMBA.>")








>")

 Can’t solve originalCan solve relaxed PRMs sample randomly but… start goal C-obst difficult to sample points.>")

 Immune-system-inspired.>")





