Download presentation
Presentation is loading. Please wait.

1
Lecture 8: Plutonium Chemistry
From: Chemistry of actinides Nuclear properties and isotope production Pu in nature Separation and Purification Atomic properties Metallic state Compounds Solution chemistry

2
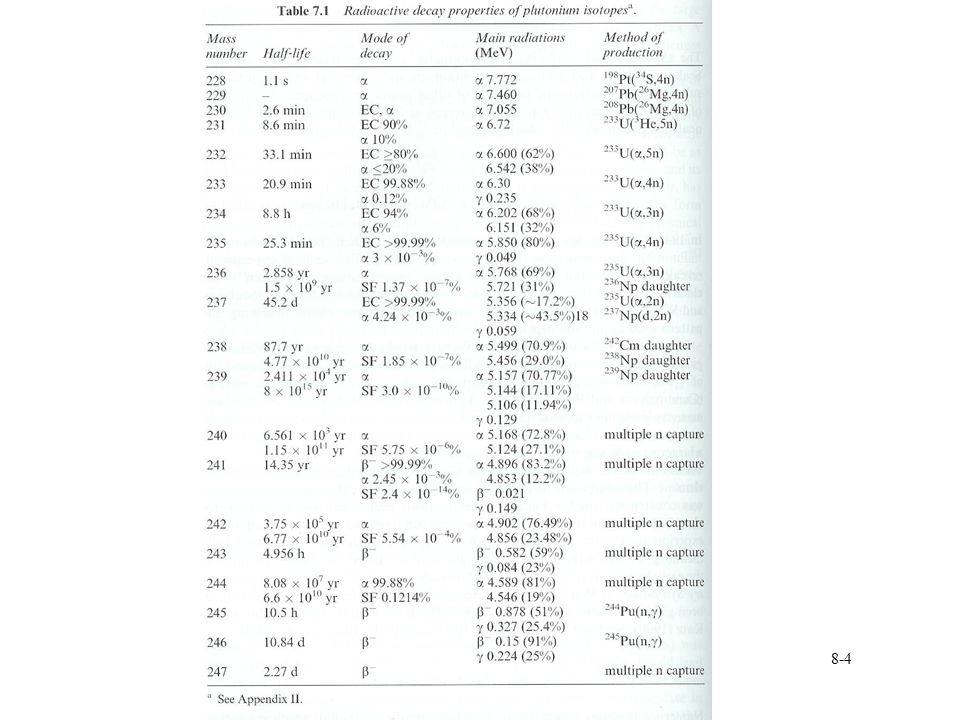
Pu nuclear properties Isotopes from 228≤A≤247 Important isotopes 238Pu
237Np(n,g)238Np 238Pu from beta decay of 238Np Separated from unreacted Np by ion exchange Decay of 242Cm 0.57 W/g Power source for space exploration 83.5 % 238Pu, chemical form as dioxide Enriched 16O to limit neutron emission 6000 n s-1g-1 0.418 W/g PuO2 150 g PuO2 in Ir-0.3 % W container
238Np. 238Pu from beta decay of 238Np. Separated from unreacted Np by ion exchange. Decay of 242Cm W/g. Power source for space exploration % 238Pu, chemical form as dioxide. Enriched 16O to limit neutron emission n s-1g W/g PuO g PuO2 in Ir-0.3 % W container.")
3
Pu nuclear properties 239Pu 2.2E-3 W/g
Basis of formation of higher Pu isotopes Pu first from nuclear test Higher isotopes available Longer half lives suitable for experiments

5
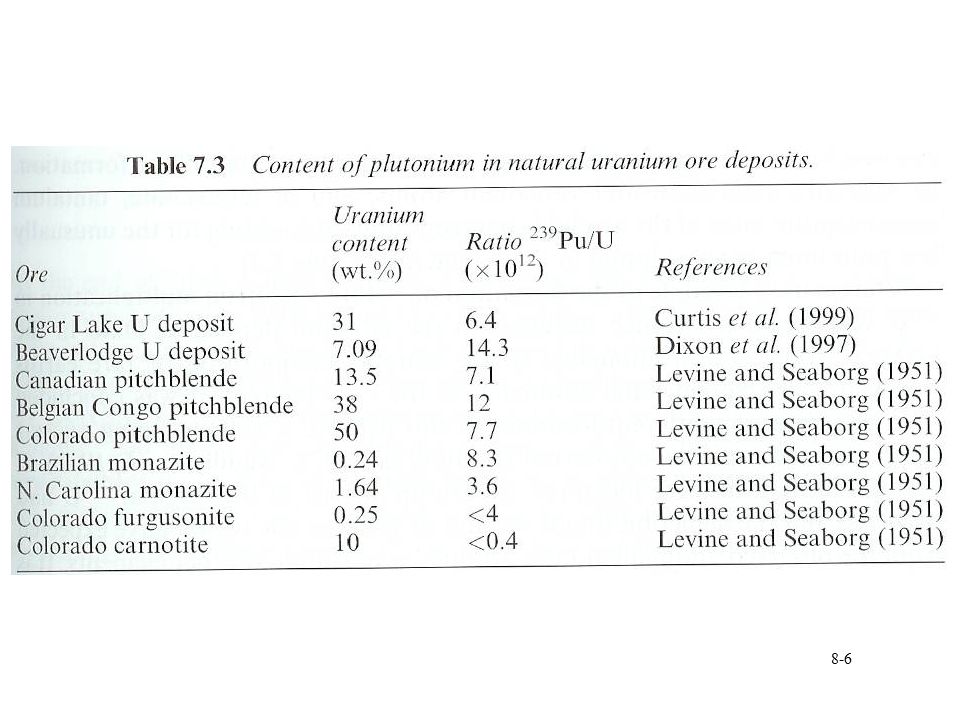
Pu in nature Most Pu due to anthropogenic sources
239,244Pu can be found in nature 239Pu from nuclear processes occurring in U ore n,g reaction Neutrons from SF of U neutron multiplication in 235U a,n on light elements 24.2 fission/g U/hr, need to include neutrons from 235U 244Pu Based on Xe isotopic ratios SF of 244Pu 1E-18 g 244Pu/g bastnasite mineral

7
Pu separations 1855 MT Pu produced Current rate of 70-75 MT/years
225 MT for fuel cycle 260 MT for weapons Large scale separations based on manipulation of Pu oxidation state Aqueous (PUREX) Non-aqueous (Pyroprocessing) Precipitation methods Basis of bismuth phosphate separation Precipitation of BiPO4 in acid carries tri- and tetravalent actinides Bismuth nitrate and phosphoric acid Separation of solid, then oxidation to Pu(VI) Sulfuric acid forms solution U sulfate, preventing precipitation Used after initial purification methods LaF3 for precipitation of trivalent and tetravalent actinides
 Non-aqueous (Pyroprocessing) Precipitation methods. Basis of bismuth phosphate separation. Precipitation of BiPO4 in acid carries tri- and tetravalent actinides. Bismuth nitrate and phosphoric acid. Separation of solid, then oxidation to Pu(VI) Sulfuric acid forms solution U sulfate, preventing precipitation. Used after initial purification methods. LaF3 for precipitation of trivalent and tetravalent actinides.")
9
Pu separations Solvent extraction
Some novel chemistry with third phase formation

10
Pu separations Extraction chromatography Extractant on solid support
Ion-exchange Both cation and anion exchange Anion exchange based on formation of appropriate species in acidic solution Change of solution impact sorption to column Pu separation Sorb Pu(IV,VI) in 6 M acid, reduce to Pu(III)
 in 6 M acid, reduce to Pu(III)")
11
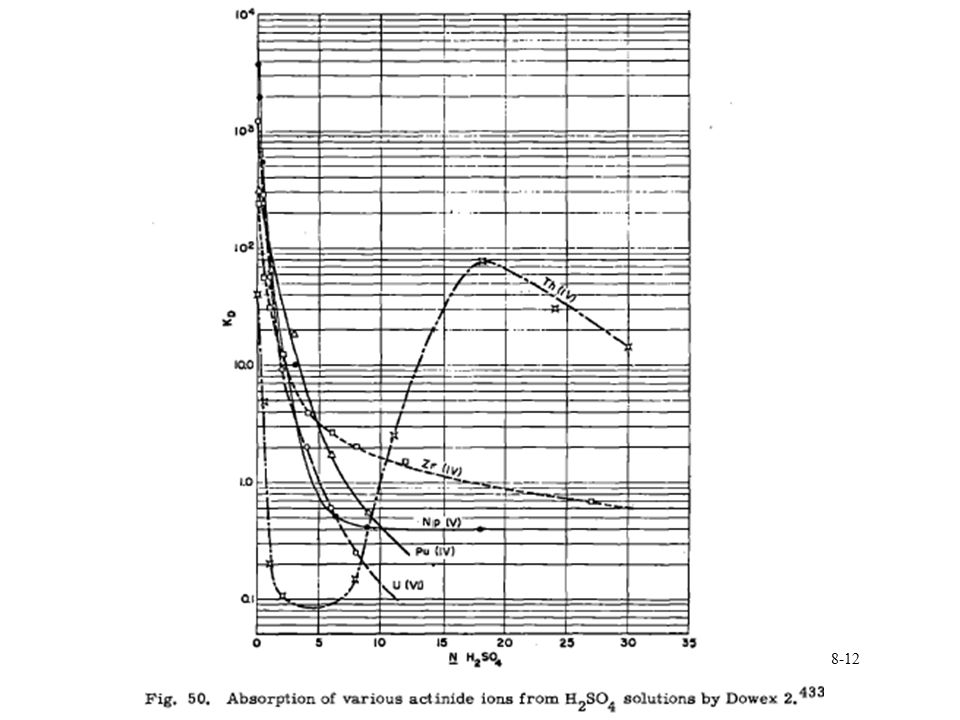
Pu anion exchange

14
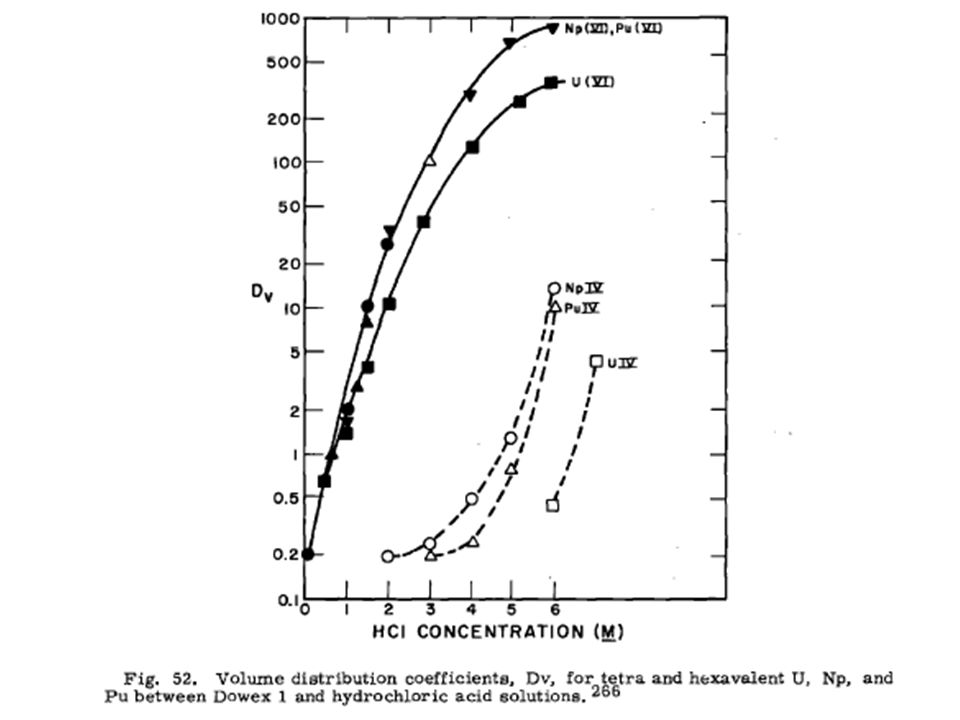
Pu cation exchange General cation exchange trends for Pu
HN03, H2S04, and HC104 show stronger influence than HC1 Strong increase in distribution coefficient in HClO4 at high acidities exhibited for Pu(III) and Pu(VI)
 and Pu(VI)")
16
Pu separations Alkaline solutions
Need strong ligands that can compete with hydroxide to form different species F-, CO32-, H2O2 High solubility, based on oxidation state Stabilize Pu(VII) Room temperature ionic liquids Quaternary ammonium with anions AlCl4-, PF6- Liquid-liquid extraction Electrochemical disposition N S O C F 3
 Room temperature ionic liquids. Quaternary ammonium with anions. AlCl4-, PF6- Liquid-liquid extraction. Electrochemical disposition. N. S. O. C. F. 3.")
17
Pu separations Halide volatility (PuF6, PuCl6)
PuO2 in fluidized bed reactor with fluorine at 400° C Can substitute NH4HF2 for some fluorination Also use of O2F2 PuF6 decomposes to PuF4 and F2 in a thermal decomposition column Supercritical fluid extraction Most research with CO2 Use complexants dissolved in SCF TBP.HNO3, TTA for extraction from soil Change of pressure to achieve separations

18
Pu atomic properties Ground state configuration [Rn]5f67s2
Term symbol 7F0 Optical emission Pu I spectra Within 3 eV (24000 cm-1) 5f56d7s2, 5f66d7s, 5f56d27s, 5f67s7p, 5f57s27p, and 5f56d7s7p Large number of electronic states and thousands of spectral lines Isotopic influence on spectra
![Pu atomic properties Ground state configuration [Rn]5f67s2](http://slideplayer.com/slide/6612420/23/images/18/Pu+atomic+properties+Ground+state+configuration+%5BRn%5D5f67s2.jpg "Term symbol 7F0. Optical emission Pu I spectra. Within 3 eV (24000 cm-1) 5f56d7s2, 5f66d7s, 5f56d27s, 5f67s7p, 5f57s27p, and 5f56d7s7p. Large number of electronic states and thousands of spectral lines. Isotopic influence on spectra.")
19
Pu atomic properties Moessbauer spectroscopy 238,239,240Pu
238Np beta decay, 44 keV photon 239Np beta decay, 57.3 keV photon Alpha decay of 244Cm, 42.9 keV photon

20
Metallic Pu Interests in processing-structure-properties relationship
Reactions with water and oxygen Impact of self-irradiation Density g·cm−3 Liquid density at m.p. 16.63 g·cm−3 Melting point 912.5 K Boiling point 3505 K Heat of fusion 2.82 kJ·mol−1 Heat of vaporization 333.5 kJ·mol−1 Heat capacity (25 °C) 35.5 J·mol−1·K−1
 35.5 J·mol−1·K−1.")
21
Preparation of Pu metal
Ca reduction Pyroprocessing PuF4 and Ca metal Conversion of oxide to fluoride Start at 600 ºC goes to 2000 ºC Pu solidifies at bottom of crucible Direct oxide reduction Direct reduction of oxide with Ca metal PuO2, Ca, and CaCl2 Molten salt extraction Separation of Pu from Am and lanthanides Oxidize Am to Am3+, remains in salt phase MgCl2 as oxidizing agent Oxidation of Pu and Am, formation of Mg Reduction of Pu by oxidation of Am metal

22
Pu metal Electrorefining Liquid Pu oxidizes from anode ingot into
molten salt electrode 740 ºC in NaCl/KCl with MgCl2 as oxidizing agent Oxidation to Pu(III) Addition of current causes reduction of Pu(III) at cathode Pu drips off cathode
 Addition of current causes reduction of Pu(III) at cathode. Pu drips off cathode.")
23
Pu metal Zone refining (700-1000 ºC)
Purification from trace impurities Fe, U, Mg, Ca, Ni, Al, K, Si, oxides and hydrides Melt zone passes through Pu metal at a slow rate Impurities travel in same or opposite direction of melt direction Vacuum distillation removes Am Application of magnetic field levitates Pu

24
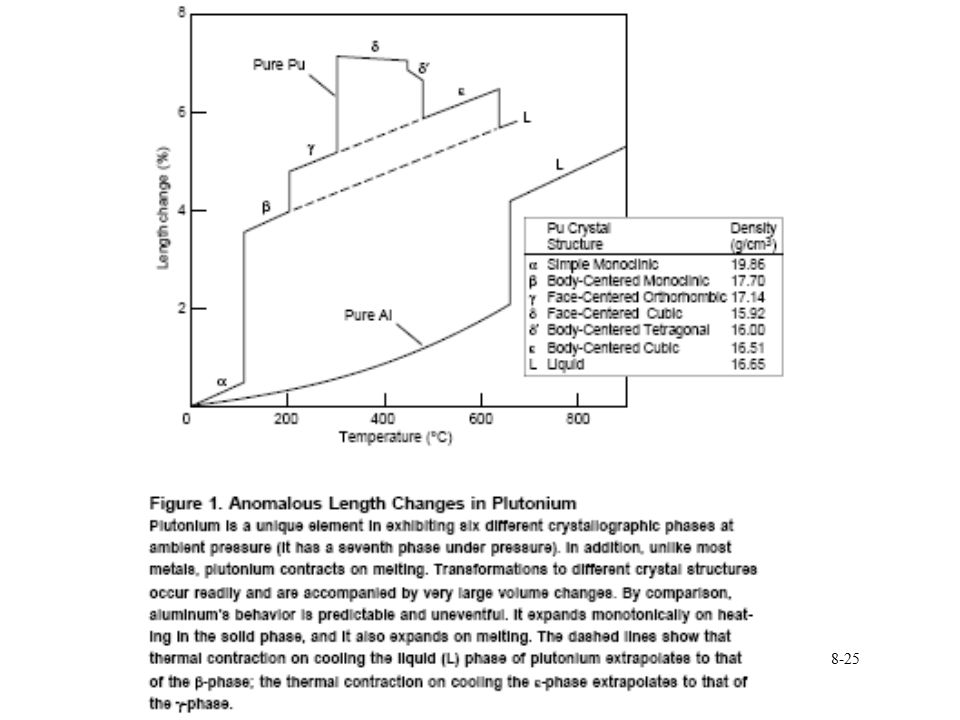
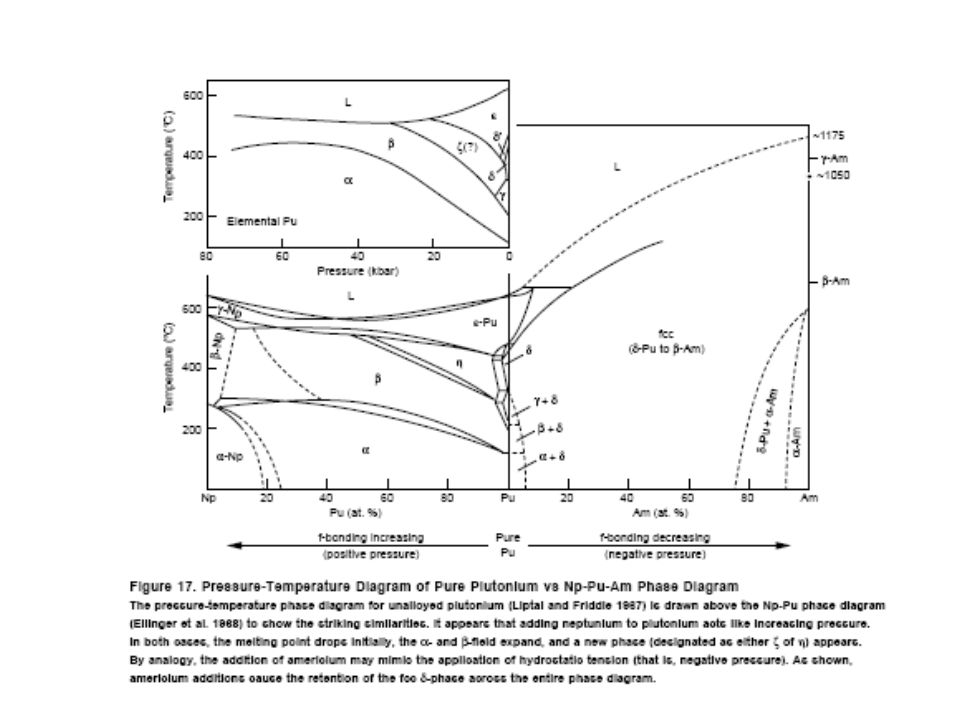
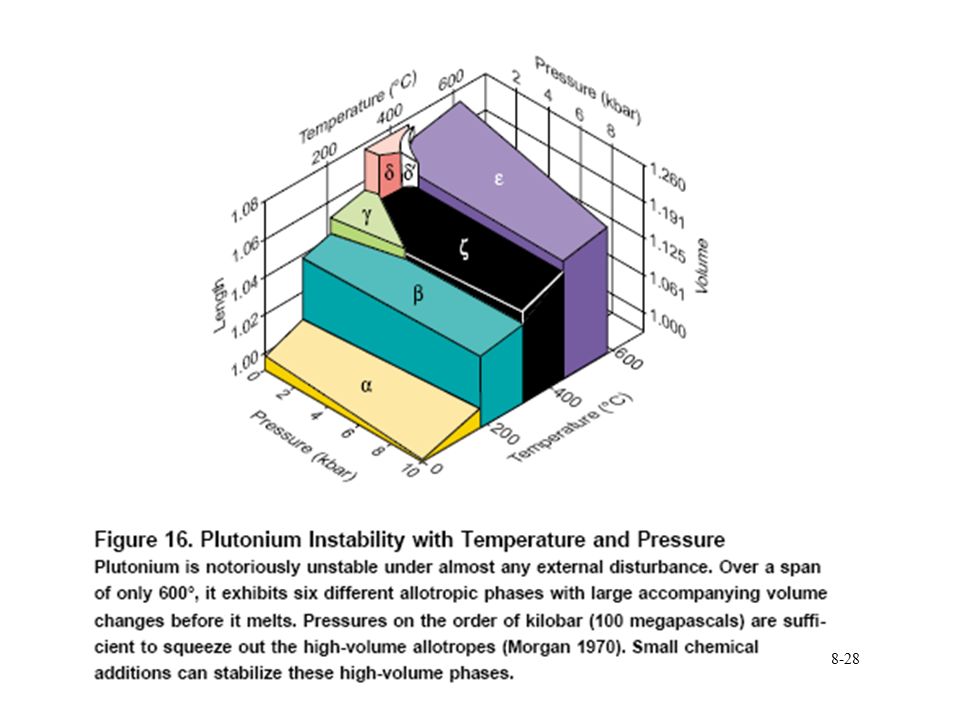
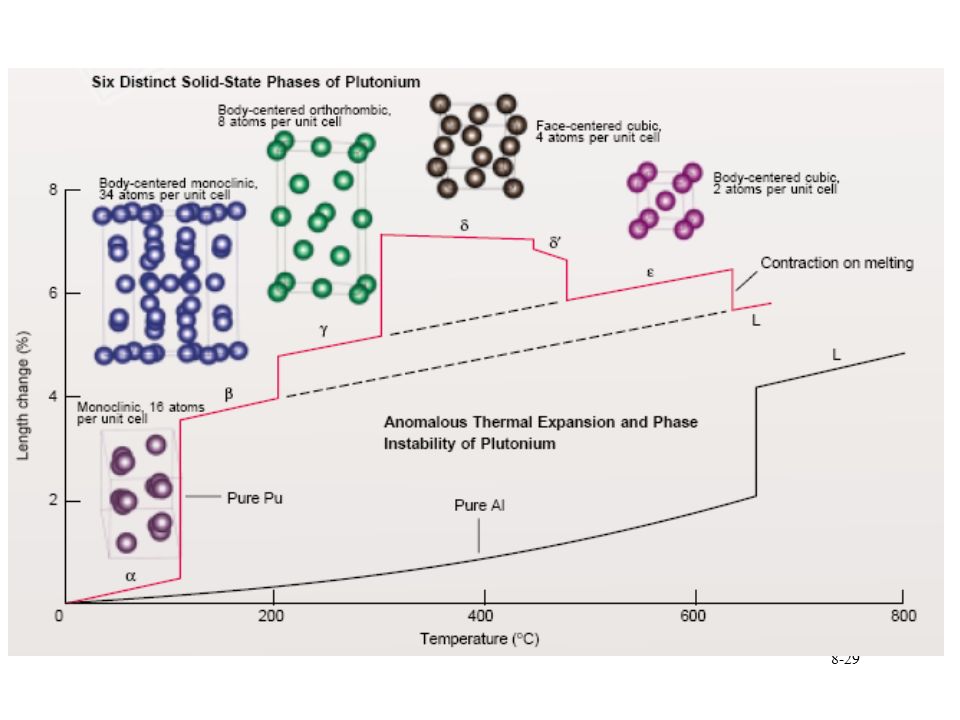
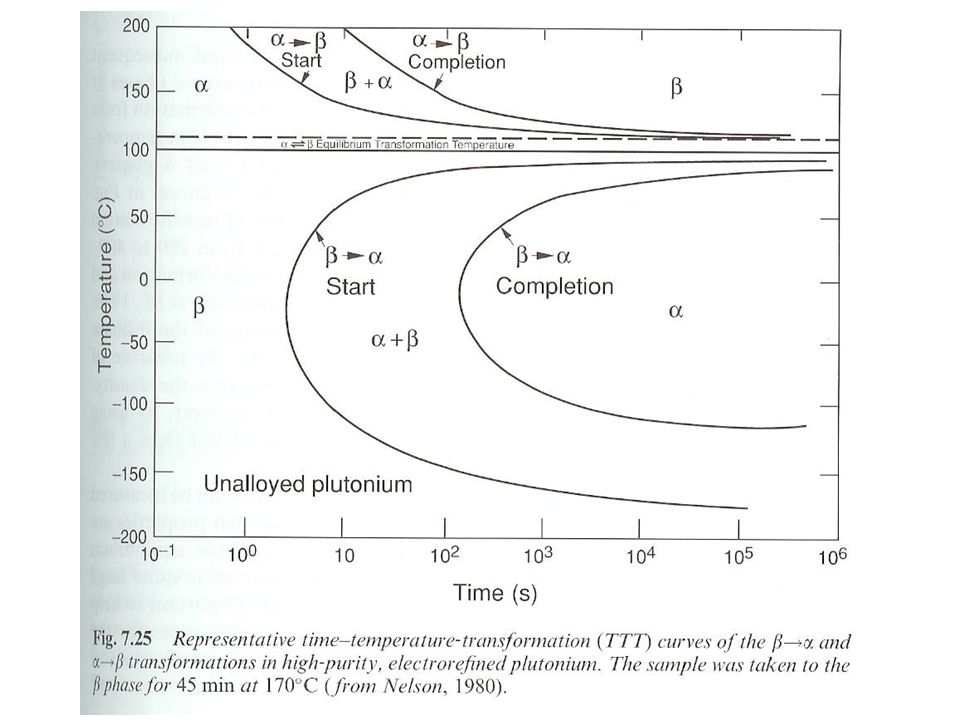
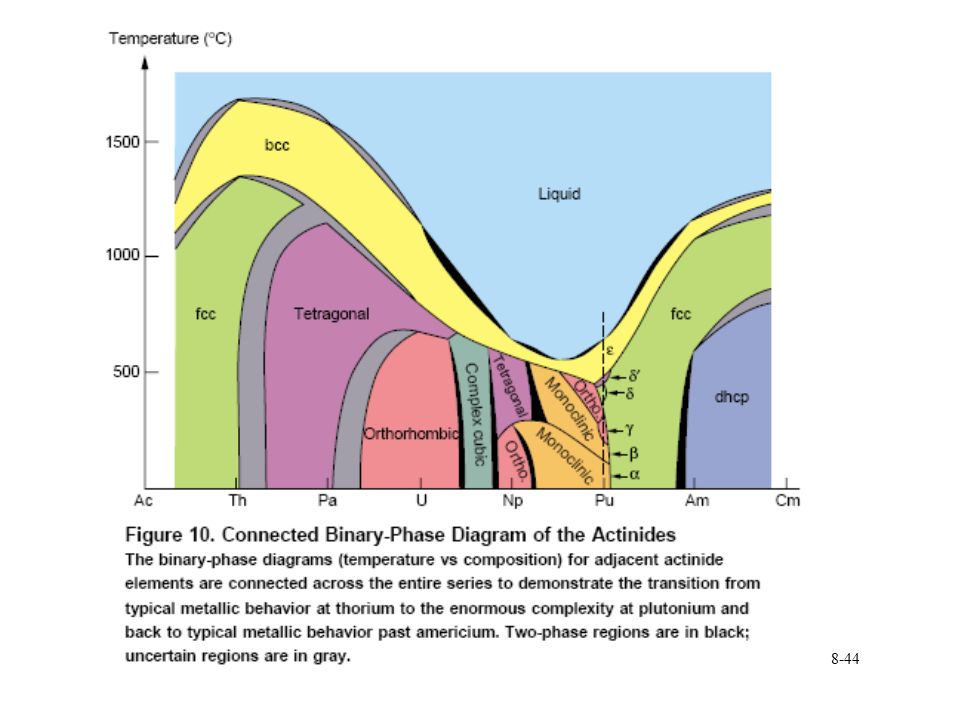
Pu phase stability 6 different Pu solid phases
7th phase at elevated pressure fcc phase least dense Energy levels of allotropic phases are very close to each other Pu extremely sensitive to changes in temperature, pressure, or chemistry Densities of the allotropes vary significantly dramatic volume changes with phase transitions Crystal structure of the allotropes closest to room temperature are of low symmetry more typical of minerals than metals. Pu expands when it solidifies from a melt Low melting point Liquid Pu has very large surface tension with highest viscosity known near the melting point. Pu lattice is very soft vibrationally and very nonlinear

30
Pu metal phases Low symmetry ground state for a phase due to 5f bonding Higher symmetry found in transition metals f orbitals have odd symmetry Basis for low symmetry (same as p orbitals Sn, In, Sb, Te) odd-symmetry p orbitals produce directional covalent-like bonds and low-symmetry noncubic structures Recent local density approximation (LDA) electronic-structure calculations show narrow width of f bands leads to low-symmetry ground states of the actinides Bandwidths are a function of volume. narrower for large volumes
 odd-symmetry p orbitals produce directional covalent-like bonds and low-symmetry noncubic structures. Recent local density approximation (LDA) electronic-structure calculations show narrow width of f bands leads to low-symmetry ground states of the actinides. Bandwidths are a function of volume. narrower for large volumes.")
31
Pu metal phase ground-state as a function of bandwidth for Nb and U and bct (body-centered tetragonal) and ort (orthorhombic), bcc (body-centered cubic) When the f band in uranium is forced to be broader than 7 eV, the high-symmetry bcc structure is stable Demonstrates narrow bands favor lower-symmetry structures for U, not that niobium true equilibrium bandwidths (Weq) are narrow (larger volumes) for the light actinides.
 and ort (orthorhombic), bcc (body-centered cubic) When the f band in uranium is forced to be broader than 7 eV, the high-symmetry bcc structure is stable. Demonstrates narrow bands favor lower-symmetry structures for U, not that niobium. true equilibrium bandwidths (Weq) are narrow (larger volumes) for the light actinides.")
32
Pu metal phase atomic-sphere approximation calculations for contributions to orbitals d fcc phase If Pu had only an f band contribution equilibrium lattice constant smaller than measured Contribution from s-p band stabilizes larger volume f band is narrow at larger volume (low symmetry) strong competition between the repulsive s-p band contribution and the attractive f band term induces instability near the ground state density-of-states functions for different low-symmetry crystal structures are very similar total energies for different low-symmetry crystal structures are very close to each other
 strong competition between the repulsive s-p band contribution and the attractive f band term induces instability near the ground state. density-of-states functions for different low-symmetry crystal structures. are very similar. total energies for different low-symmetry crystal structures are very close to each other.")
33
Pu metal phase For actinides f electron bonding increases up to Pu
Pu has the highest phase instability At Am the f electrons localize completely and become nonbonding At Am coulomb forces pull f electrons inside the valence shell leaving 2 or 3 in the s-p and d bands f-f interaction varies dramatically with very small changes in interatomic distances lattice vibrations or heating f-f and f-spd interactions with temperature may results in localization as Pu transforms from the α- to the δ-phase Low Pu melting temperature due to f-f interaction and phase instability Small temperature changes induce large electronic changes small temperature changes produce relatively large changes in free energy Kinetics important in phase transitions

35
Pu metallic radii based on 12 coordinate and extrapolated to room temperatures

36
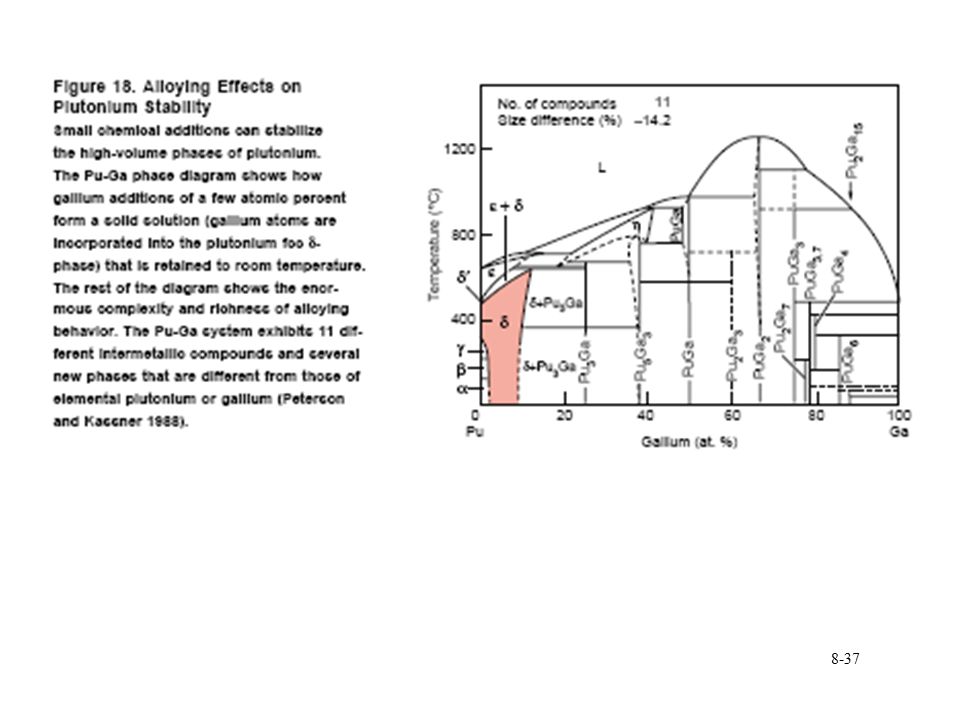
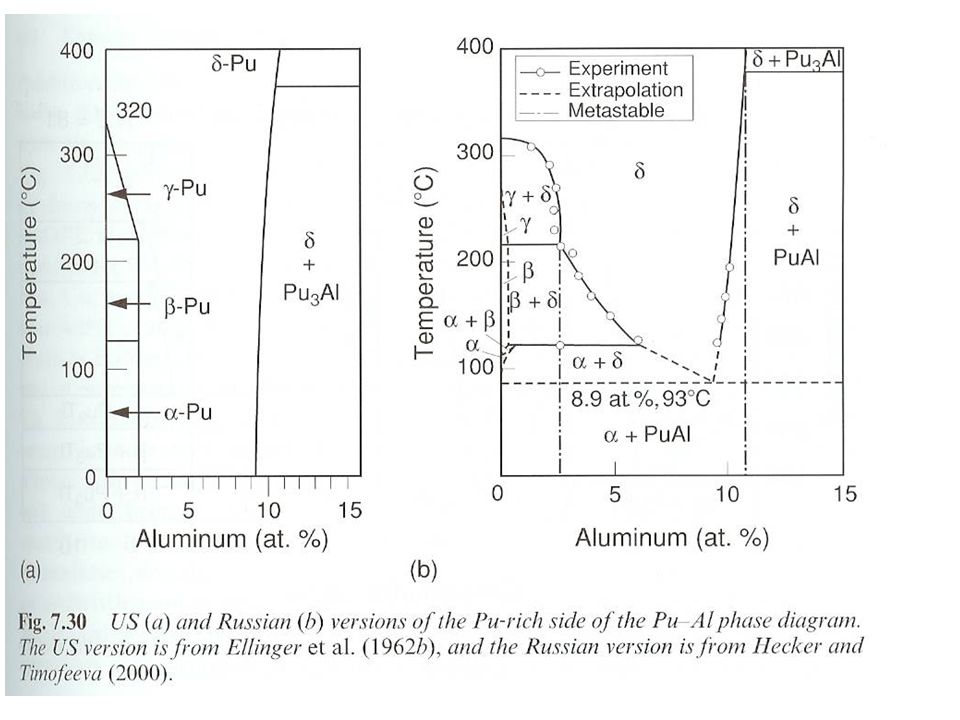
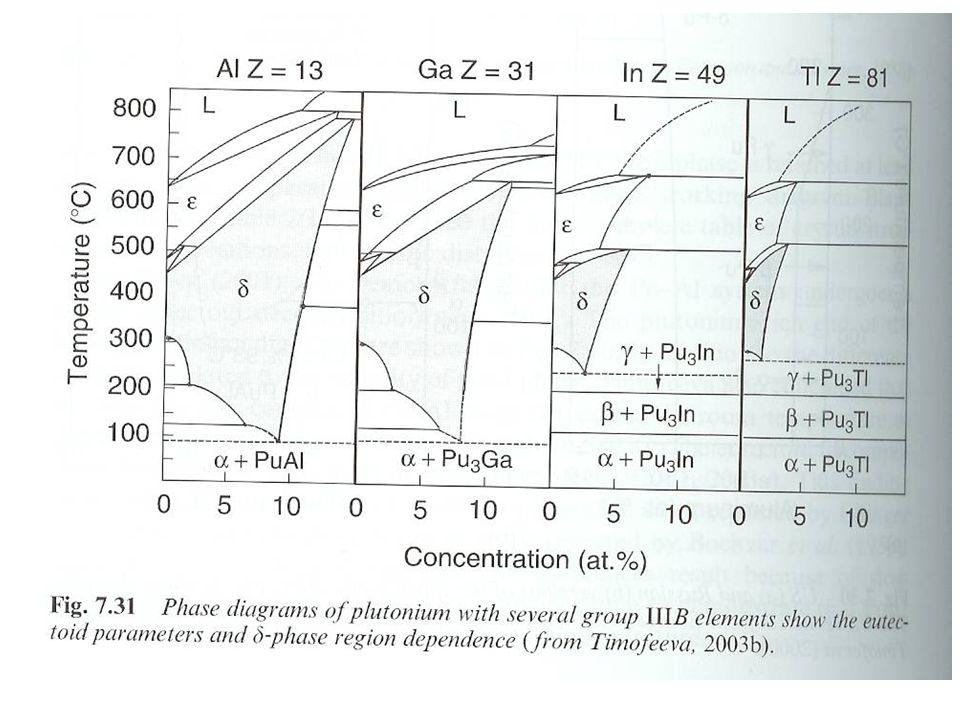
Metallic Pu Pu liquid is denser that 3 highest temperature solid phases Liquid density at g/mL Pu contracts 2.5 % upon melting Pu alloys and the d phase Ga stabilizes phase Complicated phase diagram

38
Phase never observed, slow kinetics

41
Metallic Pu Other elements that stabilize d phase
Al, Ga, Ce, Am, Sc, In, and Tl stabilize phase at room temperature Si, Zn, Zr, and Hf retain phase under rapid cooling Microstructure of d phase due to Ga diffusion in cooling Np expands the a and b phase region b phase stabilized at room temperature with Hf, Ti, and Zr Pu eutectics Pu melting point dramatically reduced by Mn Fe, Co, or Ni With Fe, mp=410 °C, 10 % Fe Used in metallic fuel Limit Pu usage (melting through cladding Interstitial compounds Large difference in ionic radii (59 %) O, C, N, and H form interstitial compounds
 O, C, N, and H form interstitial compounds.")
42
Metallic Pu Electronic structure shows competition between itinerant and localized behavior Boundary between magnetic and superconductivity 5f electrons 2 to 4 eV bands, strong mixing Polymorphism Solid state instability Catalytic activity Isolated Pu 7s25f6, metallic Pu 7s26d15f5 Lighter than Pu, addition f electron goes into conducting band Starting at Am f electrons become localized Increase in atomic volume

43
Metallic Pu Modeling to determine electronic structure and bonding properties Density functional theory Describes an interacting system of fermions via its density not via the many-body wave function 3 variables (x,y,z) rather than 3 for each electron For actinides need to incorporate Low symmetry structures Relativistic effects Electron-electron correlations local-density approximation (LDA) Include external potential and Coulomb interactions approximation based upon exact exchange energy for uniform electron gas and from fits to the correlation energy for a uniform electron gas Generalized gradient approximation (GGA) Localized electron density and density gradient Total energy calculations at ground state
 rather than 3 for each electron. For actinides need to incorporate. Low symmetry structures. Relativistic effects. Electron-electron correlations. local-density approximation (LDA) Include external potential and Coulomb interactions. approximation based upon exact exchange energy for uniform electron gas and from fits to the correlation energy for a uniform electron gas. Generalized gradient approximation (GGA) Localized electron density and density gradient. Total energy calculations at ground state.")
45
Modeling Pu metal electronic configuration
Pu metal configuration 7s26d15f5 From calculations, all eight valence electrons are in the conduction band, 5f electrons in α-plutonium behave like the 5d electrons of the transition metals than the 4f of the lanthanides Bonding and antibonding orbitals from sum and differences of overlapping wavefunctions Complicated for actinides Small energy difference between orbital can overlap in solids Accounts for different configurations

46
bandwidth narrows with increasing orbital angular momentum
Larger bands increase probability of electrons moving d and f electrons interact more with core electrons Narrowing reflects decreasing radial extent of orbitals with higher angular momentum, or equivalently decrease in overlap between neighboring atoms Enough f electrons in Pu to be significant Relativistic effects are important

47
Transition at Pu

48
For Pu, degree of f electron localization varies with phase

49
5f electrons extend relatively far from the nucleus compared to the 4f electrons
5f electrons participate in chemical bonding much-greater radial extent of the probability densities for the 7s and 7p valence states compared with 5f valence states 5f and 6d radial distributions extend farther than shown by nonrelativistic calculations 7s and 7p distributions are pulled closer to the ionic cores in relativistic calculations

50
Pu metal physical and thermodynamic properties
Already reviewed density and thermal expansion Heat capacity Difficulties in measurement due to self-heating and damage 239Pu 2.2 mW/g Low temperature measurements do not permit annealing Use of 242Pu helps overcome decay related issues

51
The Specific Heat of Plutonium and Other Metals
The low-temperature specific heat of a metal is the sum of a lattice term an electronic term In this figure, the line for copper represents the behavior of most metals whereas the lines for α- and δ-plutonium have the highest values of γ (intercept values) of any pure element Indicating that conduction electrons have an enhanced effective mass The compound UBe13 has an extremely high electronic specific heat, which continues to increase until it is cut off by the compound’s transition to superconductivity just below 1 K The superconductivity of UBe13 proves that its large heat capacity must be associated with the conduction electrons
 of any pure element. Indicating that conduction electrons have an enhanced effective mass. The compound UBe13 has an extremely high electronic specific heat, which continues to increase until it is cut off by the compound’s transition to superconductivity just below 1 K. The superconductivity of UBe13 proves that its large heat capacity must be associated with the conduction electrons.")
52
Pu metal magnetic behavior
Magnetic susceptibility (c) Internal response of material to applied magnetic field M = χB, M is magnetization of the material B is the magnetic field intensity Large values for Pu and alloys
 Internal response of material to applied magnetic field. M = χB, M is magnetization of the material. B is the magnetic field intensity. Large values for Pu and alloys.")
53
Susceptibilities of Pu higher than most metals,
lower than materials with local moment Variation in susceptibility as plutonium changes phase increase slightly as temperature decreases

55
Pu metal mechanical properties
Related to crystal structure and melting point Pu has a range of structures with different melting points Results in a variety of mechanical properties Stress, oxidation, corrosion, pyrophoricity, self-irradiation Sensitive to chemistry (alloying) and processing (microstructure)
 and processing (microstructure)")
56
Pu metal mechanical properties
Stress/strain properties High strength properties bend or deform rather than break Beyond a limit material abruptly breaks Fails to absorb more energy

57
Pu metal mechanical properties
α-plutonium is strong and brittle, similar to cast iron elastic response with very little plastic flow Stresses increase to point of fracture strength of the unalloyed α-phase decreases dramatically with increasing temperature Similar to bcc and hcp metals. Pu-Ga δ-phase alloys show limited elastic response followed by extensive plastic deformation low yield strength ductile fracture

58
For α-Pu elastic limit is basically fracture strength
The Pu-Ga alloy behaves more like Al Fails by ductile fracture after elongation

59
Tensile-test results for unalloyed Pu
Related to temperature and resulting change in phases Strengths of α- and β-phase are very sensitive to temperature Less pronounced for γ-phase and δ-phase data represent work of several investigators different purity materials, and different testing rates Accounts for variations in values, especially for the α-Pu phase

60
Pu metal mechanical properties
Metal elastic response due to electronic structure and resulting cohesive forces Metallic bonding tends to result in high cohesive forces and high elastic constants Metallic bonding is not very directional since valence electrons are shared throughout the crystal lattice Results in metal atoms surrounding themselves with as many neighbors as possible close-packed, relatively simple crystal structures The Pu 5f electrons have narrow conduction bands and high density-of-states energetically favorable for ground-state crystal structure to distort to low-symmetry structures at room temperature Pu has typical metal properties at elevated temperatures or in alloys

61
Pu metal corrosion and oxidation
Formation of oxide layer Can include oxides other than dioxide Slow oxidation in dry air Greatly enhanced oxidation rate in presence of water or hydrogen Metal has pyrophoric properties Corrosion depends on chemical condition of Pu surface Pu2O3 surface layer forms in absence or low amounts of O2 Promotes corrosion by hydrogen Pu hydride (PuHx, where 1.9 < x < 3) increases oxidation rate in O2 by 1013 PuO2+x surface layer forms on PuO2 in the presences of water enhances bulk corrosion of Pu metal in moist air
 increases oxidation rate in O2 by PuO2+x surface layer forms on PuO2 in the presences of water. enhances bulk corrosion of Pu metal in moist air.")
62
O2 sorbs on Pu surface to form oxide layer
Oxidation continues but O2 must diffuse through oxide layer Oxidation occurs at oxide/metal interface Oxide layer thickness initially increases with time based on diffusion limitation At oxide thickness around 4–5 μm in room temperature surface stresses cause oxide particles to spall oxide layer reaches a steady-state thickness further oxidation and layer removal by spallation Eventually thickness of oxide layer remains constant

63
Steady state in dry air at room temperature
steady-state layer of Pu2O3 at oxide-metal interface Pu2O3 thickness is small compared with the oxide thickness at steady state Autoreduction of dioxide by the metal at the oxide metal interface produces Pu2O3 Pu2O3 reacts with the diffusing O2 to form dioxide

64
Arrhenius Curves for Oxidation of Unalloyed and Alloyed Plutonium in Dry Air and Water Vapor
ln of the reaction rate R versus 1/T slope of each curve is proportional to the activation energy for the corrosion reaction Curve 1 oxidation rate of unalloyed plutonium in dry air or dry O2 at a pressure of 0.21 bar. Curve 2a increase in the oxidation rate when unalloyed metal is exposed to water vapor up to 0.21 bar, equal to the partial pressure of oxygen in air Curves 2b and 2c show the moisture-enhanced oxidation rate at water vapor pressure of 0.21 bar in temperature ranges of 61°C–110°C and 110°C–200°C, respectively Curves 1’ and 2’ oxidation rates for the δ-phase gallium-stabilized alloy in dry air and moist air (water vapor pressure ≤ 0.21 bar), respectively Curve 3 transition region between the convergence of rates at 400°C and the onset of the autothermic reaction at 500°C Curve 4 temperature-independent reaction rate of ignited metal or alloy under static conditions rate is fixed by diffusion through an O2-depleted boundary layer of N2 at the gas-solid interface Curve 5 temperature-dependent oxidation rate of ignited droplets of metal or alloy during free fall in air
, respectively. Curve 3 transition region between the convergence of rates at 400°C and the onset of the autothermic reaction at 500°C. Curve 4 temperature-independent reaction rate of ignited metal or alloy under static conditions. rate is fixed by diffusion through an O2-depleted boundary layer of N2 at the gas-solid interface. Curve 5 temperature-dependent oxidation rate of ignited droplets of metal or alloy during free fall in air.")
65
Oxide Layer on Plutonium Metal under Varying Conditions
corrosion rate is strongly dependent on the metal temperature varies significantly with the isotopic composition,quantity, geometry, and storage configuration steady-state oxide layer on plutonium in dry air at room temperature (25°C) is shown at the top (a) Over time, isolating PuO2-coated metal from oxygen in a vacuum or an inert environment turns the surface oxide into Pu2O3 by the autoreduction reaction At 25°C, the transformation is slow time required for complete reduction of PuO2 depends on the initial thickness of PuO2 layer highly uncertain because reaction kinetics are not quantified above 150°C, rapid autoreduction transforms a several micrometer-thick PuO2 layer to Pu2O3 within minutes (b) Exposure of the steady-state oxide layer to air results in continued oxidation of the metal Kinetic data indicate that a one-year exposure to dry air at room temperature increases the oxide thickness by about 0.1 μm At a metal temperature of 50°C in moist air (50% relative humidity), the corrosion rate increases by a factor of approximately 104 corrosion front advances into unalloyed metal at a rate of 2 mm per year 150°C–200°C in dry air, the rate of the autoreduction reaction increases relative to that of the oxidation reaction steady-state condition in the oxide shifts toward Pu2O3,
 is shown at the top. (a) Over time, isolating PuO2-coated metal from oxygen in a vacuum or an inert environment turns the surface oxide into Pu2O3 by the autoreduction reaction. At 25°C, the transformation is slow. time required for complete reduction of PuO2 depends on the initial thickness of PuO2 layer. highly uncertain because reaction kinetics are not quantified. above 150°C, rapid autoreduction transforms a several micrometer-thick PuO2 layer to Pu2O3 within minutes. (b) Exposure of the steady-state oxide layer to air results in continued oxidation of the metal. Kinetic data indicate that a one-year exposure to dry air at room temperature increases the oxide thickness by about 0.1 μm. At a metal temperature of 50°C in moist air (50% relative humidity), the corrosion rate increases by a factor of approximately 104. corrosion front advances into unalloyed metal at a rate of 2 mm per year. 150°C–200°C in dry air, the rate of the autoreduction reaction increases relative to that of the oxidation reaction. steady-state condition in the oxide shifts toward Pu2O3,")
66
PuO2+x Study Examined Pu oxides by two methods X-ray diffraction (XRD)
Gives information about structure Lattice parameters X-ray photoelectron spectroscopy (XPS) Used to evaluate binding of oxygen Examined reaction of PuO2 with H2O from 25 °C to 350 °C
 Used to evaluate binding of oxygen. Examined reaction of PuO2 with H2O from 25 °C to 350 °C.")
67
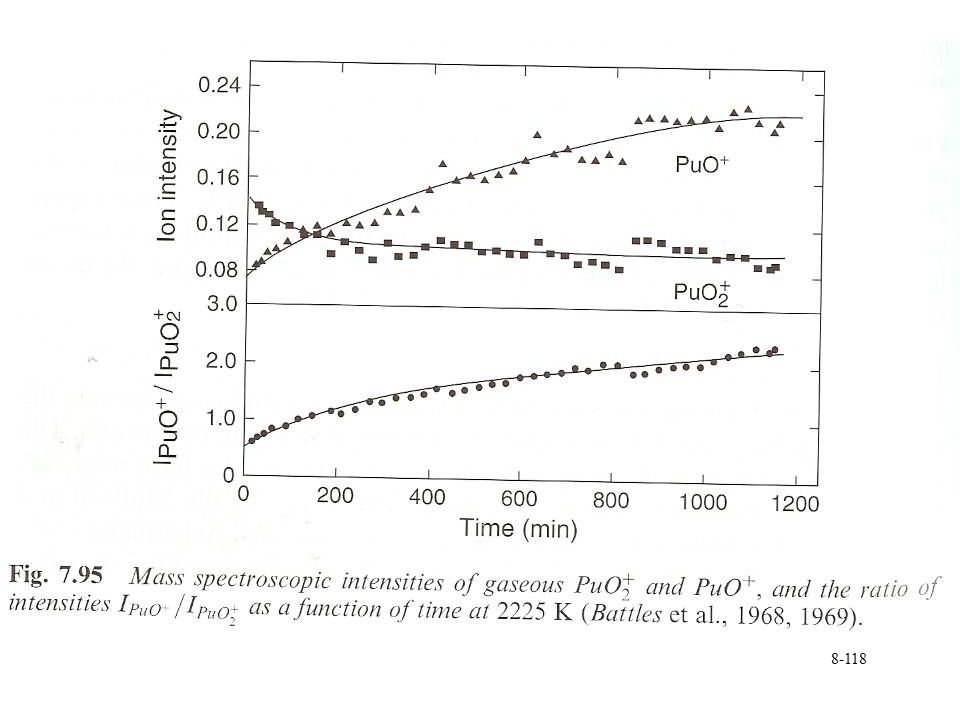
Results Mass spectrometric analysis shows production of H2(g)
PuO2(s)+xH2O(abs) <--> PuO2+x(s)+ xH2(g) 350°C H2 pressure formation 300°C 250°C 200°C Time (hr)
+xH2O(abs) <--> PuO2+x(s)+ xH2(g) 350°C. H2 pressure formation. 300°C. 250°C. 200°C. Time (hr)")
68
Results Lattice parameter change Attributed to increase in Pu:O ratio
Cubic lattice parameter variation ao= (O:Pu) Relative insensitivity attributed to formation of Pu(VI) Extra O forms plutonyl O/Pu ratio
 Relative insensitivity attributed to formation of Pu(VI) Extra O forms plutonyl. O/Pu ratio.")
69
Results X-ray photoelectric spectroscopy data
High binding energies for the oxide 442 eV, 429 eV 4f5/2, 4f7/2 Pu(VI) or Pu(VII) No Pu(V) O 1s spectrum consistent with oxide Absence of OH- attributed to continued reaction of water
 or Pu(VII) No Pu(V) O 1s spectrum consistent with oxide. Absence of OH- attributed to continued reaction of water.")
70
Results PuO2+x formed via catalytic cycle
Driven by H2O sorbed to surface If O2 present, H reforms water Formation of water drives catalytic cycle

71
Pu oxide coating reaction with H2
Plutonium hydride (PuHx) fcc phase forms a continuous solid solution for 1.9 < x < 3.0 Pu(s) + (x/2)H2(g) → PuHx(s) observed value of x depends on hydrogen pressure and temperature hydride is readily oxidized by air decomposes back to its component elements when heated in continuously pumped vacuum Hydriding occurs only after the ubiquitous dioxide layer on the metal is penetrated Unlike oxidation the reaction of hydrogen initiates at a limited number of nucleation sites a single nucleation site typically appears only after a lengthy, but unpredictable, induction period Once formed sites are the most reactive areas of the surface Hydriding rate is proportional to the active area covered by the hydride Increases exponentially over time to a maximum value as sites grow and ultimately cover the surface . At that point, the rate Temperatures between –55°C and 350°C and a H2 pressure of 1 bar reaction at a fully active surface consumes plutonium at a constant rate of 6–7 g/cm2 min Advances into metal or alloy at about 20 cm/h
 fcc phase. forms a continuous solid solution for 1.9 < x < 3.0. Pu(s) + (x/2)H2(g) → PuHx(s) observed value of x depends on hydrogen pressure and temperature. hydride is readily oxidized by air. decomposes back to its component. elements when heated in continuously pumped vacuum. Hydriding occurs only after the ubiquitous dioxide layer on the metal is penetrated. Unlike oxidation the reaction of hydrogen initiates at a limited number of nucleation sites. a single nucleation site typically appears only after a lengthy, but unpredictable, induction period. Once formed sites are the most reactive areas of the surface. Hydriding rate is proportional to the active area covered by the hydride. Increases exponentially over time to a maximum value as sites grow and ultimately cover the surface. . At that point, the rate. Temperatures between –55°C and 350°C and a H2 pressure of 1 bar. reaction at a fully active surface consumes plutonium at a constant rate of 6–7 g/cm2 min. Advances into metal or alloy at about 20 cm/h.")
72
Rates for Catalyzed Reactions of Pu with H2, O2, and Air
Diffusion-limited oxidation data shown in gray compared to data for the rates of reactions catalyzed by surface compounds oxidation rates of PuHx-coated metal or alloy in air the hydriding rates of PuHx- or Pu2O3-coated metal or alloy at 1 bar of pressure, oxidation rates of PuHx-coated metal or alloy in O2 rates are extremely rapid, values are constant indicate the surface compounds act as catalysts

73
Hydride-Catalyzed Oxidation of Pu
After the hydride-coated metal or alloy is exposed to O2, oxidation of the pyrophoric PuHx forms a surface layer of oxide and heat H2 formed by the reaction moves into and through the hydride layer to reform PuHx at the hydride-metal interface sequential processes in reaction oxygen adsorbs at the gas-solid interface as O2 O2 dissociates and enters the oxide lattice as an anionic species thin steady-state layer of PuO2 may exist at the surface oxide ions are transported across the oxide layer to the oxide-hydride interface oxide may be Pu2O3 or PuO2–x (0< x <0.5 Oxygen reacts with PuHx to form heat (~160 kcal/mol of Pu) and H2 H2 produced at the oxide-hydride interface moves through the PuHx layer to the hydride-metal interface reaction of hydrogen with Pu produces PuH2 and heat
 and H2. H2 produced at the oxide-hydride interface moves. through the PuHx layer to the hydride-metal interface. reaction of hydrogen with Pu produces PuH2 and heat.")
74
rupture in inner container for Pu metal

75
increase in the inner vessel’s diameter near the ruptured end shows the extent of hydride-catalyzed corrosion during a 3-hour period

76
Radiation damage Decay rate for 239Pu is sufficient to produce radiation damage Buildup of He and radiation damage within the metal radiation damage is caused mainly by the uranium nuclei recoil energy from the decay to knock plutonium atoms from their sites in the crystal lattice of the metal Vacancies are produced Effect can produce void swelling On the microscopic level, the vacancies tend to diffuse through the metal and cluster to form voids Macroscopically, the net effect the metal swells

77
Pu Decay and the Generation of Defects
α particle has a range of about 10 μm through the Pu uranium nucleus range is only about 12 nm Both particles produce displacement damage Frenkel pairs namely vacancies and interstitial atoms Occurs predominantly at the end of their ranges Most of the damage results from the uranium nucleus and is confined to the collision cascade region

78
Stages for Radiation-Induced Void Swelling

79
Predictions for Radiation-Induced Damage in Pu
predicted contributions to volume distortion in stabilized plutonium at 70°C Distortions due to void swelling are likely to be much larger than those due to helium-bubble formation large uncertainty in the transient period prevents estimating when the void swelling should begin its linear growth rate figure shows several possible swelling curves

80
Pu Compounds Original difficulties in producing compounds Amount of Pu
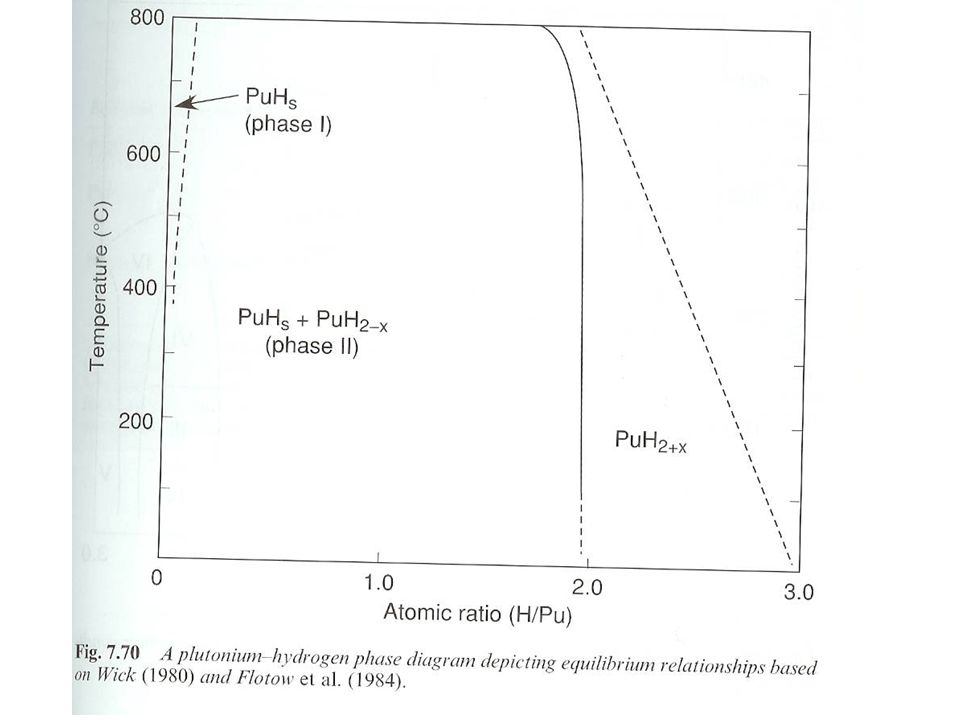
Purity Aided by advances in microsynthesis and increase in amount of available starting material Much early effort in characterization by XRD Pu Hydrides PuHx x varies from 1.9< x <3.0 Pu + x/2 H2PuHx H2 partial pressure used to control exact stoichiometry Variations and difficulties rooted in desorption of H2 Pu hydride crystallizes in a fluorite structure

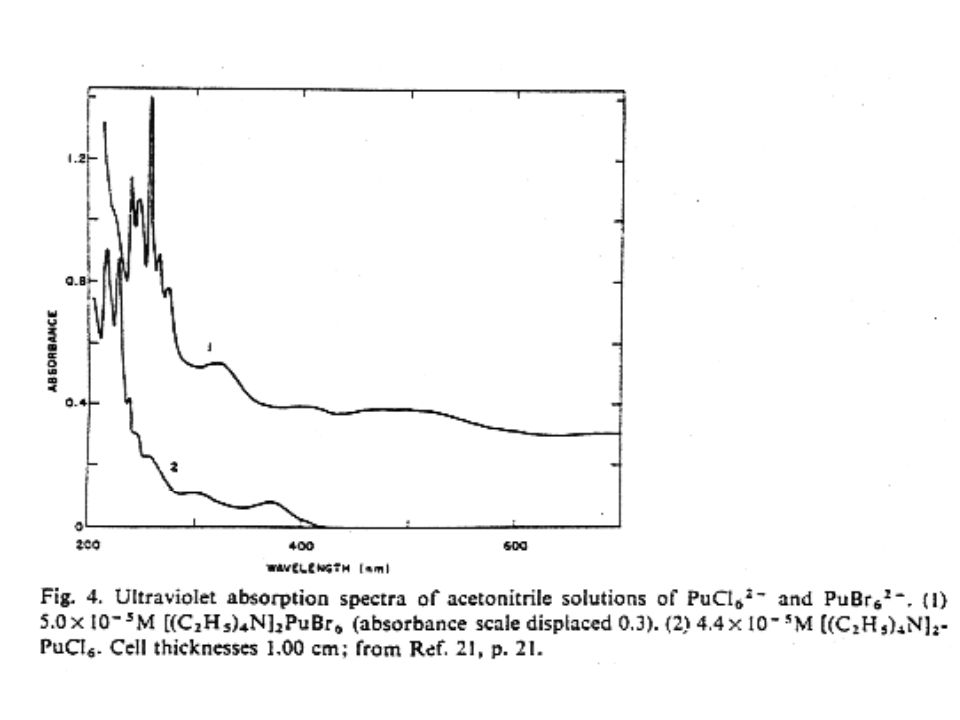
81
Pu hydride Pu hydride oxidation state
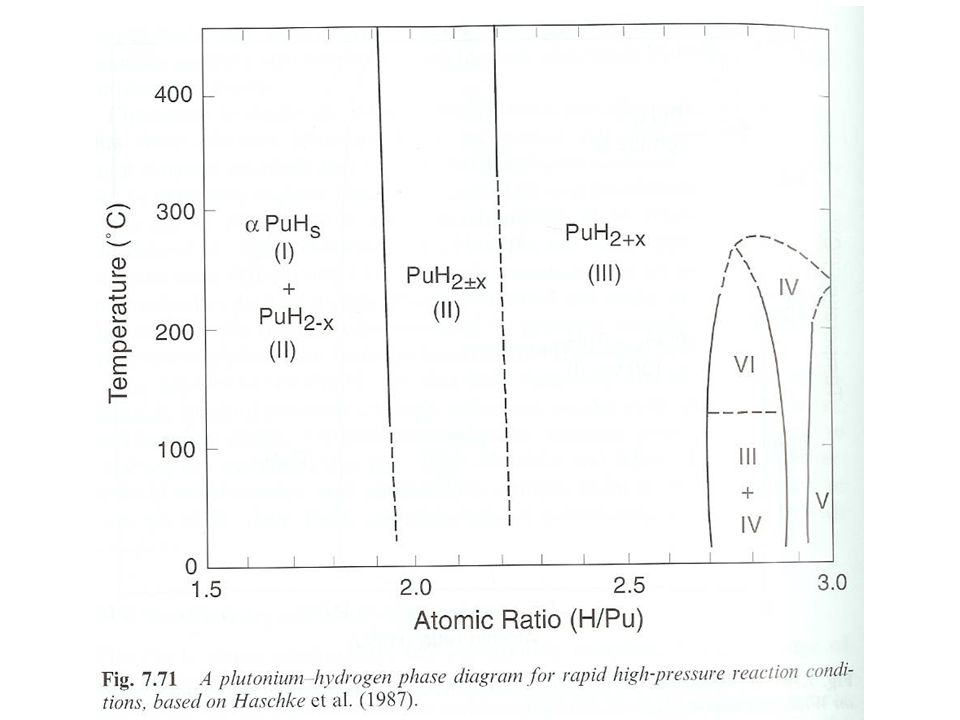
PuH2 implies divalent Pu, an unstable oxidation state Pu(II) measurements show Pu as trivalent and PuH2 is metallic Pu(III), 2 H- and 1e- Electron in conduction band Consistent with electrical conductivity measurements showing PuHx progressively changes from a metallic to semiconductor with increasing x Electrons removed from conduction band and bound as H– on octahedral sites as the hydride increases Phase relationships Two differing phase diagrams Temperature, pressure, reaction rates differ Dependence on the formation of saturated Pu hydride Hydrogen saturated Pu (PuHs) forms and co-exists with Pu hydride 5f electrons localized in Pu(III) compounds High pressure hydride synthesis results in more complex diagram Coexistence of cubic (PuH2.77) and hexagonal PuH2.88 (Region III and IV). Orthorhombic PuH2.95 (Region V) Region VI in high pressure hydride synthesis phase diagram is unknown
 measurements show Pu as trivalent and PuH2 is metallic. Pu(III), 2 H- and 1e- Electron in conduction band. Consistent with electrical conductivity measurements showing PuHx progressively changes from a metallic to semiconductor with increasing x. Electrons removed from conduction band and bound as H– on octahedral sites as the hydride increases. Phase relationships. Two differing phase diagrams. Temperature, pressure, reaction rates differ. Dependence on the formation of saturated Pu hydride. Hydrogen saturated Pu (PuHs) forms and co-exists with Pu hydride. 5f electrons localized in Pu(III) compounds. High pressure hydride synthesis results in more complex diagram. Coexistence of cubic (PuH2.77) and hexagonal PuH2.88 (Region III and IV). Orthorhombic PuH2.95 (Region V) Region VI in high pressure hydride synthesis phase diagram is unknown.")
84
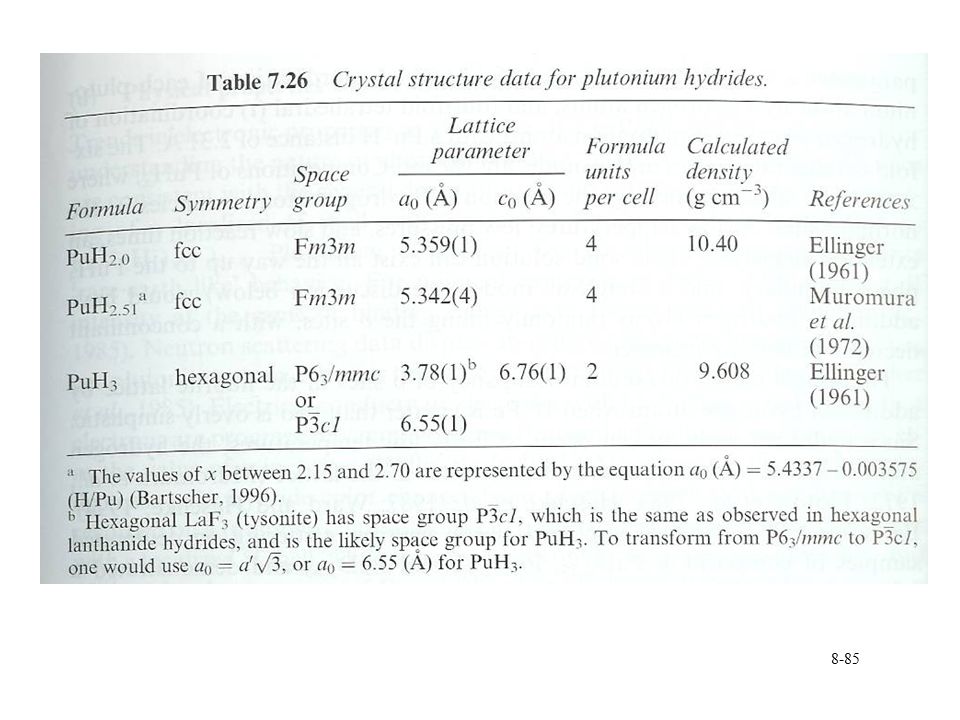
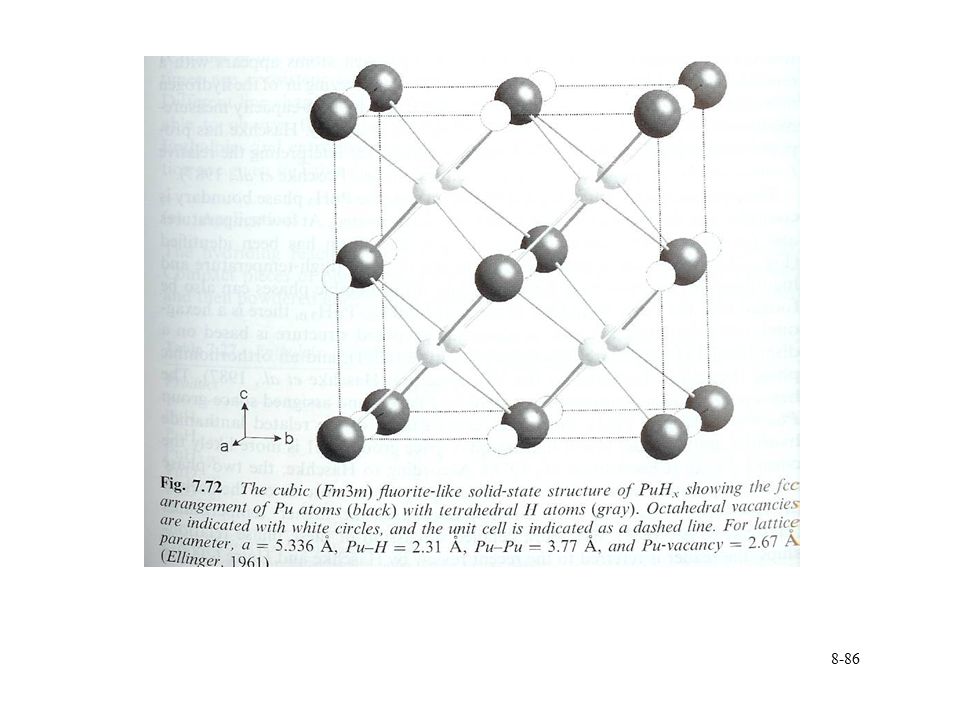
Pu hydride Solid state structure
Similar to lanthanide trifluoride system Cubic from PuHx 1.9 to 2.7 Decrease in lattice parameter with increasing x 5.36 to 5.34 Å Hexagonal beyond x=2.9 Hydrogen mobility in structure H found at octahedral and tetrahedral sites Replacement with deuterium Neutron scattering to correlate H location with stoichiometry Structure becomes complicated at x near 3

87
Aries process Hydride used to prepare metal
Formation of hydride from metal Heated to 400 °C under vacuum to release hydrogen Can convert to oxide (with O2) or nitride (N2) gas addition during heating
 or nitride (N2) gas addition during heating.")
88
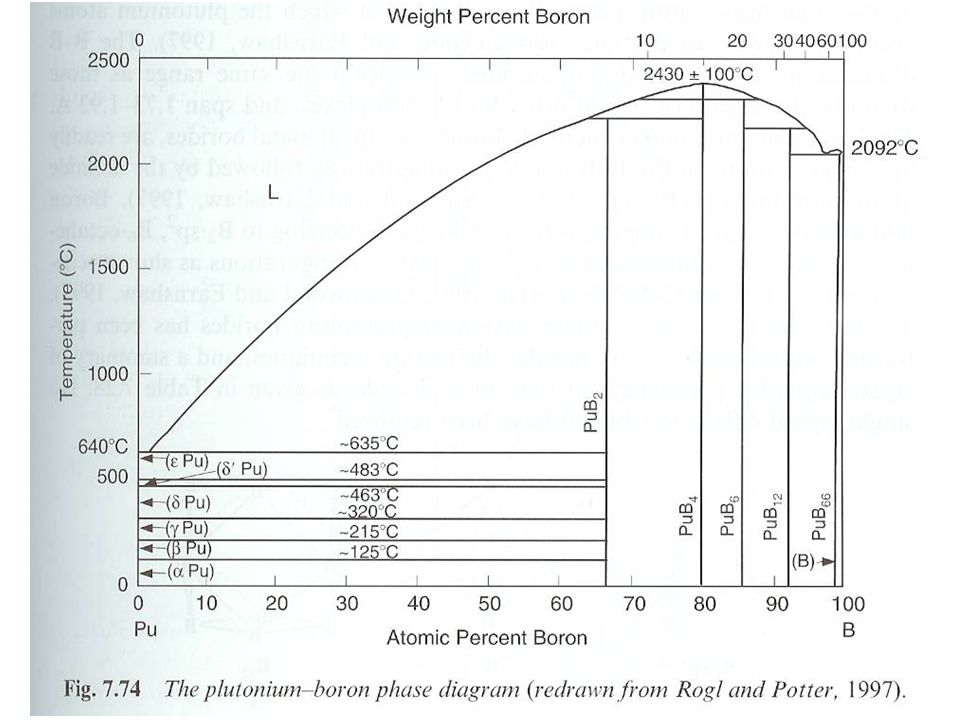
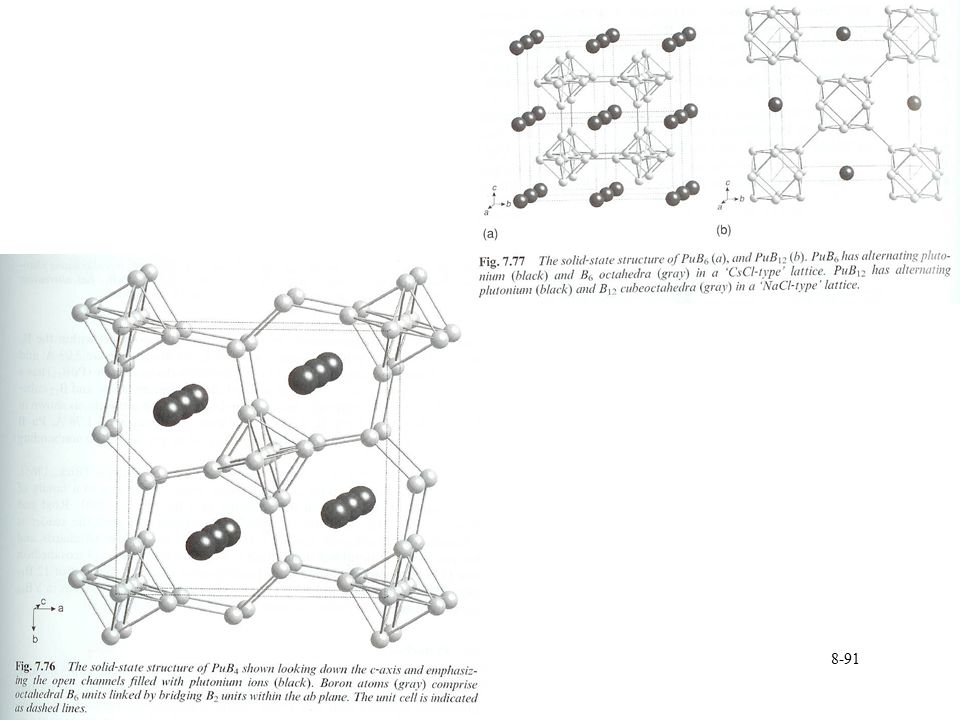
Pu borides Range of compounds PuBx x= 2, 4, 6, 12, 66
Potential storage or waste form for Pu High melting points Little work performed on compounds Prepared from heating elements Under vacuum between 900 °C and 1200 °C Arc melting under Ar Pu hydride can also act as starting material Structure Dominated by B-B bonding Similar to most metal borides Pu occupies vacant sites Power XRD, no single crystal data Little data on properties Some data on magnetic properties Suggest tetravalent Pu for diborides Based on comparison to Np complexes

92
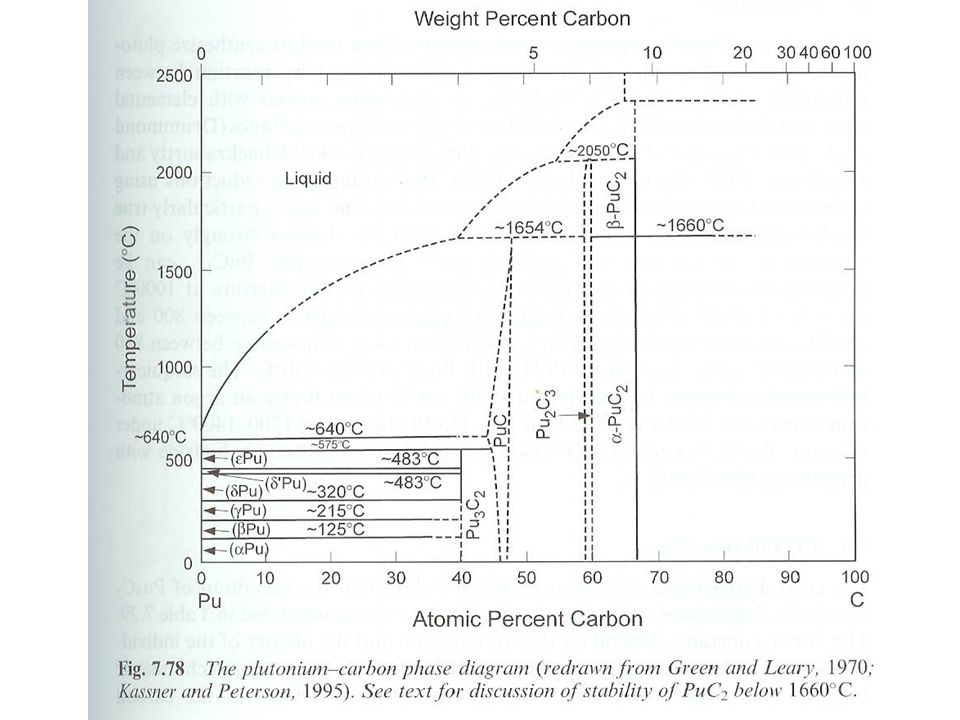
Pu carbides Four known compounds Pu3C2, PuC1-x, Pu2C3, and PuC2
PuC exists only as substoichiometric compound PuC0.6 to PuC0.92 Compound considered candidate for fuels Synthesis At high temperatures elemental C with: Pu metal Pu hydrides Pu oxides Oxygen impurities present with oxide starting material High Pu carbides can be used to produce other carbides PuC1-x from PuH2 and Pu2C3 at 700 °C Final product composition dependent upon synthesis temperature, atmosphere (vacuum or Ar) and time
 and time.")
93
Pu carbides Structure Lattice constant depends upon composition
As C content increases, C are replaced by C2 units Pu2C3 from Pu4(C2)3 Cubic structure with 12 C2 units in cell Studied by XRD and neutron scattering XRD not accurate for C Neutron scattering shows C-C of Å in Pu2C3 C2 bond in acetylene is 1.20 Å PuC2 Variation of XRD data with temperature High temperature (1710 °C) cubic Room temperature tetragonal unit cell
3. Cubic structure with 12 C2 units in cell. Studied by XRD and neutron scattering. XRD not accurate for C. Neutron scattering shows C-C of Å in Pu2C3. C2 bond in acetylene is 1.20 Å. PuC2. Variation of XRD data with temperature. High temperature (1710 °C) cubic. Room temperature tetragonal unit cell.")
94
Pu carbides Chemical properties
PuC1-x oxidizes in air starting at 200 °C Slower reaction with N2 Formation of PuN at 1400 °C Pu2C3 has reactions similar to PuC1-x All Pu carbides dissolve in HNO3-HF mixtures Liberation of CO2 with oxidizing acids With lower carbides formation of other organics Mellitic and oxalic acids Thermodynamic properties PuC1-x evaporates upon heating Ternary phases prepared Pu-U-C M3C2, MC1-x, M2C3, and MC2 observed Pu-Th-C Mixed carbide-nitrides, carbide-oxides, and carbide hydrides have been prepared

96
Pu-silicon system Five known Pu-Si compounds
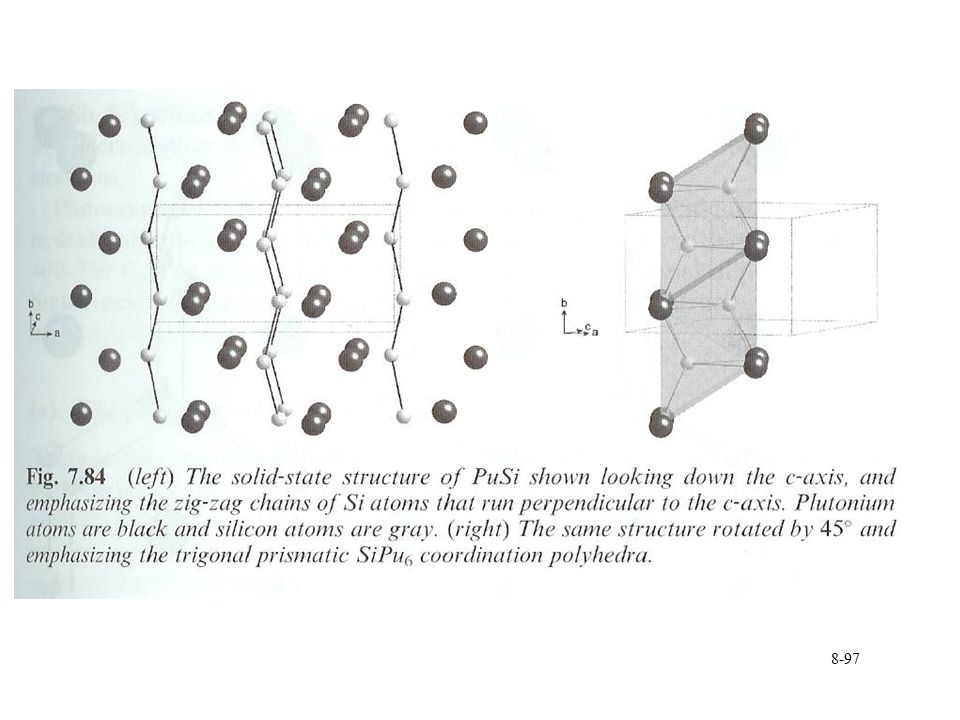
5:3, 3:2, 1:1, 3:5, and 1:2 (Pu:Si) Highest melting point for 3:5 at 1646 °C Synthesis Reaction with PuF3 at 1200 °C under vacuum 4 PuF3 + (3+4x)Si4 PuSix + 3 SiF4 SiF4 is volatile and removed Arc melting of Si and Pu or PuHx under Ar PuO2 with Si or SiC at 1400 °C under vacuum Structures Commonalities with borides Production of isolated Si2 units or structures (chains, layers, networks) with increased Si content Properties Metallic appearance Pyrophoric Oxidize in air to form PuO2 Reacts with water High melting point and high densities 8.96 g/cm3 (Pu3Si5) g/cm3 (PuSi) 11.98 g/cm3 (Pu5Si3)
 Highest melting point for 3:5 at 1646 °C. Synthesis. Reaction with PuF3 at 1200 °C under vacuum. 4 PuF3 + (3+4x)Si4 PuSix + 3 SiF4. SiF4 is volatile and removed. Arc melting of Si and Pu or PuHx under Ar. PuO2 with Si or SiC at 1400 °C under vacuum. Structures. Commonalities with borides. Production of isolated Si2 units or structures (chains, layers, networks) with increased Si content. Properties. Metallic appearance. Pyrophoric. Oxidize in air to form PuO2. Reacts with water. High melting point and high densities g/cm3 (Pu3Si5) g/cm3 (PuSi) g/cm3 (Pu5Si3)")
99
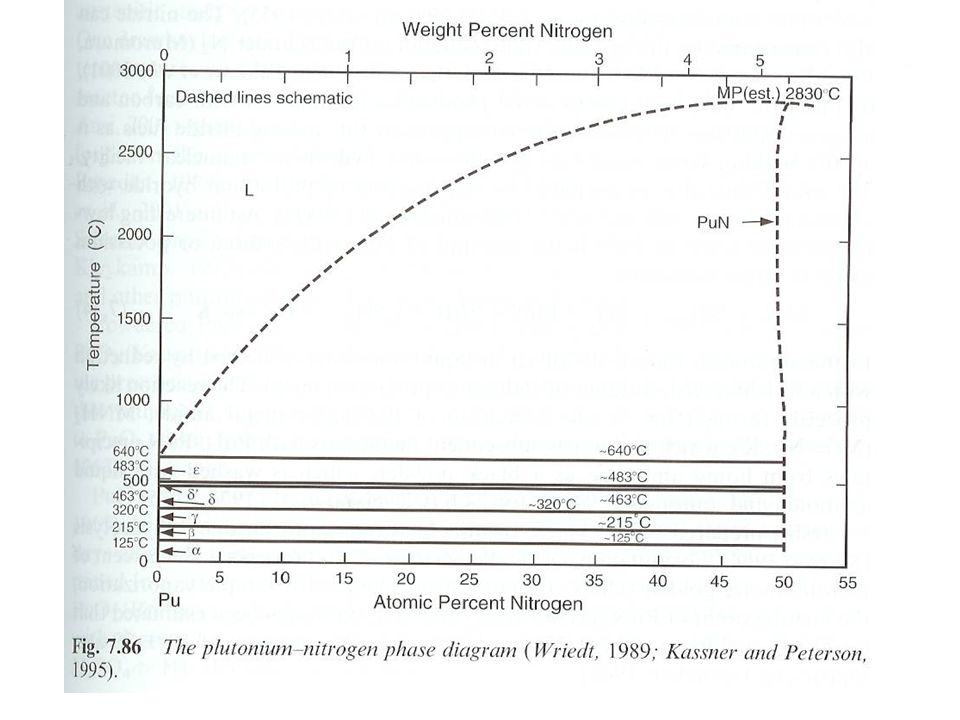
Pu pnictides Basic compounds Highest order PuX2
Prepared by reaction of Pu metal or hydride in sealed quartz tube at °C Pu-nitrogen system Only PuN known with certainty Narrow composition range Liquid Pu forms at 1500 °C PuN melting point not observed Preparation Pu hydride with N2 between 500 °C and 1000 °C Can react metal, but conversion not complete Formation in liquid ammonia PuI3 + NH3 +3 M+ PuN + 3 MI+ 1.5 H2 Intermediate metal amide MNH2 formation Pu precipitates

101
Pu nitride Structure fcc cubic NaCl structure Lattice 4.905 Å
Data variation due to impurities, self-irradiation Pu-N 2.45 Å Pu-Pu 3.47 Å Properties High melting point (estimated at 2830 °C) Compatible with steel (up to 600 °C) and Na (890 °C, boiling point) Reacts with O2 at 200 °C Reaction rates increase with H2O vapor Dissolves in mineral acids Rapidly with HNO3 Moderately delocalized 5f electrons Increases with atomic number of ligand Behavior consistant with f5 (Pu3+) Supported by correlated spin density calculations
 Compatible with steel (up to 600 °C) and Na (890 °C, boiling point) Reacts with O2 at 200 °C. Reaction rates increase with H2O vapor. Dissolves in mineral acids. Rapidly with HNO3. Moderately delocalized 5f electrons. Increases with atomic number of ligand. Behavior consistant with f5 (Pu3+) Supported by correlated spin density calculations.")
102
Pu-P system Formed from Pu hydride with PH3 at elevated temperature
Pu hydride with excess red phosphorus in pressure vessel at °C under Ar Excess P removed by distillation at 300 °C Melts with decomposition at 2600 °C Pu As and Pu Sb compounds also form Pu4Sb3 formed in addition to mono species

103
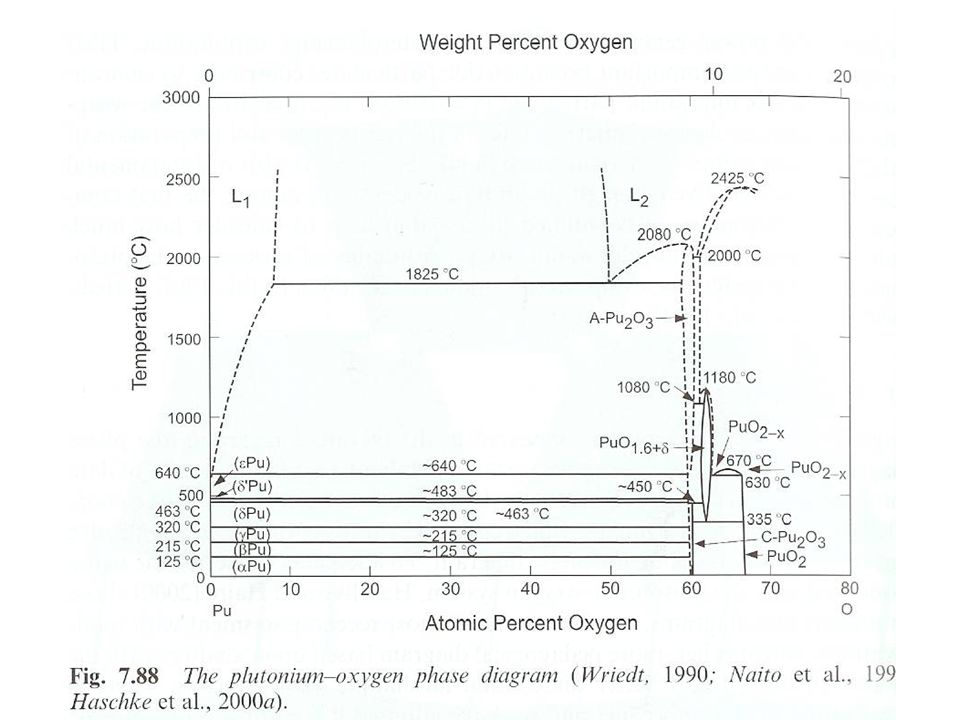
Pu oxide Pu storage, fuel, and power generators Important species
Corrosion Environmental behavior Different Pu oxide solid phases PuO Pu2O3 Composition at 60 % O Different forms at PuOx x=1.52, bcc x=1.61, bcc PuO2 fcc, wide composition range (1.6 <x<2)
")
105
Pu oxide preparation PuO Existence of phase uncertain
Definitely identified in gas phase IR spectrum Not indicated in phase diagram Surface film on Pu metal Molten Pu metal with stoichiometric Ag2O Reduction of PuO2 with C at °C Reduction of PuOCl or PuO2 with Ba vapor PuO reacts violently with O2 Some discussion on PuO actually PuOC Oxycarbide does not react violently with O2

106
Pu oxide preparation Pu2O3 Hexagonal (A-Pu2O3) and cubic (C-Pu2O3)
Distinct phases that can co-exist No observed phase transformation Kinetic behavior may influence phase formation of cubic phase C-Pu2O3 forms on PuO2 of d-stabilied metal when heated to °C under vacuum Metal and dioxide fcc, favors formation of fcc Pu2O3 Requires heating to 450 °C to produce hexagonal form Not the same transition temperature for reverse reaction Indication of kinetic effect Formed by reaction of PuO2 with Pu metal, dry H2, or C A-Pu2O3 formed PuO2+Pu2Pu2O3 at 1500 °C in Ta crucible Excess Pu metal removed by sublimation 2PuO2+CPu2O3 + CO

107
Pu oxide preparation Hyperstoichiometric sesquioxide (PuO1.6+x)
Requires fast quenching to produce of PuO2 in melt Slow cooling resulting in C-Pu2O3 and PuO2-x x at 0.02 and 0.03 Substoichiometric PuO2-x From PuO1.61 to PuO1.98 Exact composition depends upon O2 partial pressure Single phase materials Lattice expands with decreasing O

109
Pu oxide preparation PuO2 Pu metal ingited in air
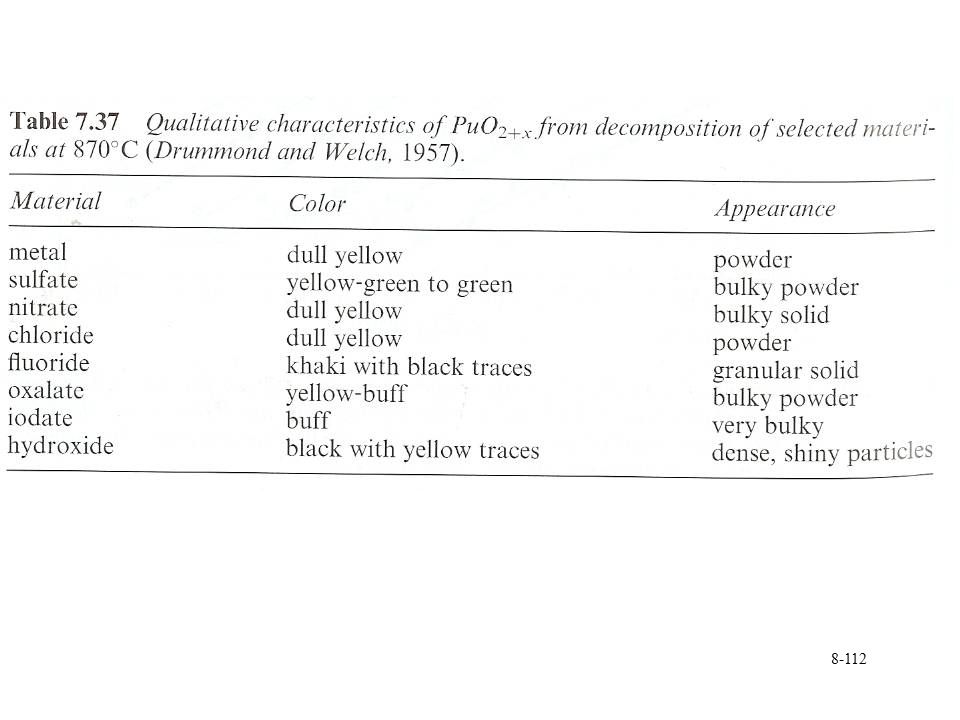
Calcination of a number of Pu compounds No phosphates Pu crystalline PuO2 formed by heating Pu(III) or Pu(IV) oxalate to 1000 °C in air Oxalates of Pu(III) forms a powder, Pu(IV) is tacky solid Rate of heating can effect composition due to decomposition and gas evolution PuO2 is olive green Can vary due to particle size, impurities Pressed and sintered for heat sources or fuel Sol-gel method Nitrate in acid injected into dehydrating organic (2-ethylcyclohexanol) Formation of microspheres Sphere size effects color
 or Pu(IV) oxalate to 1000 °C in air. Oxalates of Pu(III) forms a powder, Pu(IV) is tacky solid. Rate of heating can effect composition due to decomposition and gas evolution. PuO2 is olive green. Can vary due to particle size, impurities. Pressed and sintered for heat sources or fuel. Sol-gel method. Nitrate in acid injected into dehydrating organic (2-ethylcyclohexanol) Formation of microspheres. Sphere size effects color.")
110
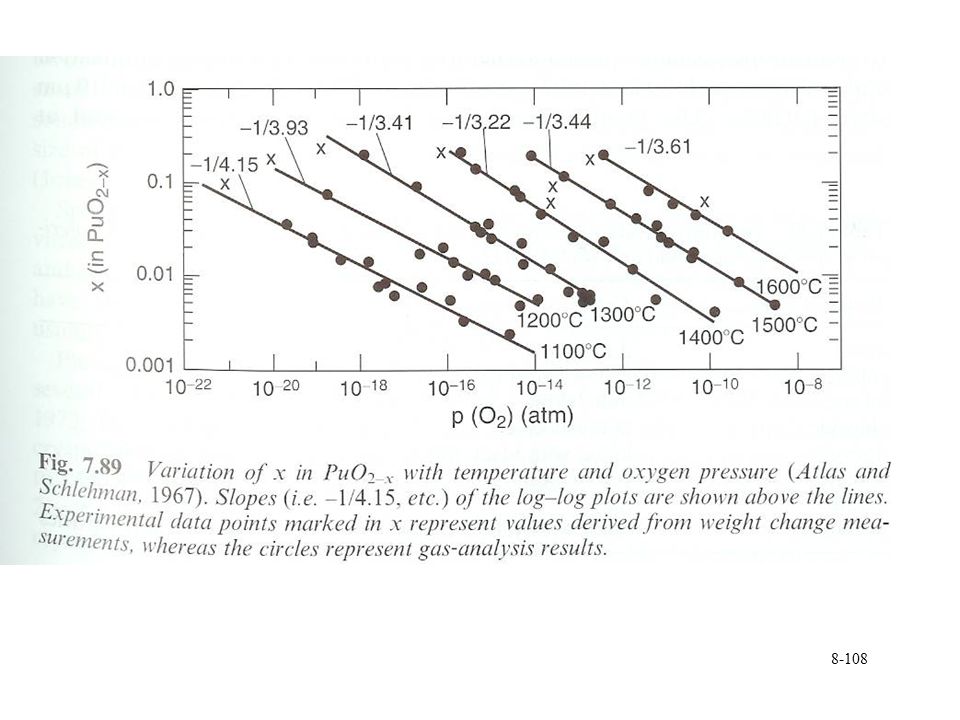
Pu oxide preparation PuO2+x, PuO3, PuO4
Tetravalent Pu oxides are favored Unable to oxidize PuO2 High pressure O2 at 400 °C Ozone PuO2+x reported in solid phase Related to water reaction PuO2+xH2OPuO2+x + xH2 Final product PuO2.3, fcc PuO3 and PuO4 reported in gas phase From surface reaction with O2 PuO4 yield decreases with decreasing O2 partial pressure

113
Pu oxide structures Lattice changes with O/Pu ratio
fcc commonality with PuO2 Related to fluorite structure

117
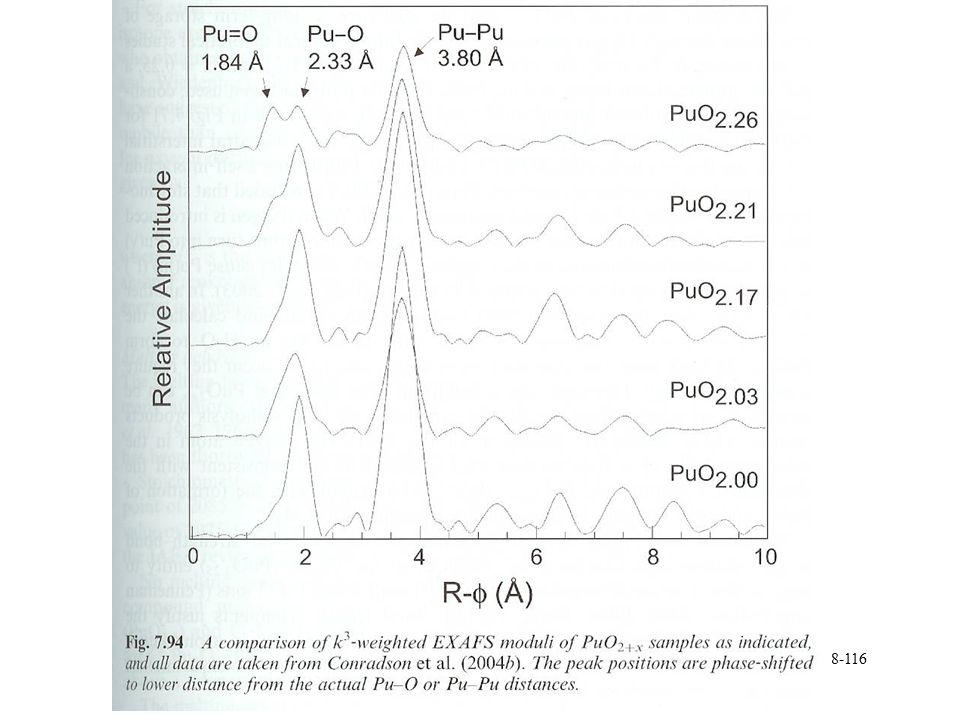
Pu oxide Oxygen at 1.84 Å Similar to Pu=O in Pu(V) complexes 1.85 Å
Interpreted as mixture of Pu(IV) and Pu(V) Oxidation by electron transfer to O f4 to f3 transition Properties Interstitial excess O and O vacancies are mobile Interstitials are more mobile Similar to O behavior in U Studied by gas phase isotope exchange Vaporization complicated Some composition change upon heating C-Pu2O3 decomposes to A-Pu2O3 and PuO1.6+x Data on vaporization conflicting Dependent upon technique, interaction with matrix PuO2 goes to PuO1.831 Gas phase PuO2+ and PuO+
 and Pu(V) Oxidation by electron transfer to O. f4 to f3 transition. Properties. Interstitial excess O and O vacancies are mobile. Interstitials are more mobile. Similar to O behavior in U. Studied by gas phase isotope exchange. Vaporization complicated. Some composition change upon heating. C-Pu2O3 decomposes to A-Pu2O3 and PuO1.6+x. Data on vaporization conflicting. Dependent upon technique, interaction with matrix. PuO2 goes to PuO Gas phase PuO2+ and PuO+")
119
Pu oxide Chemical properties
Thermodynamic parameter available for Pu oxides Dissolution High fired PuO2 difficult to dissolve Rate of dissolution dependent upon temperature and sample history Irradiated PuO2 has higher dissolution rate with higher burnup Dissolution often performed in 16 M HNO3 and 1 M HF Can use H2SiF6 or Na2SiF6 KrF2 and O2F2 also examined Electrochemical oxidation HNO3 and Ag(II) Ce(IV) oxidative dissolution
 Ce(IV) oxidative dissolution.")
120
Pu S, Se, and Te systems Forms PuX, Pu2X3, PuX2-x PuTe3
Prepared from stoichiometric reaction of PuHx and elements in sealed quartz 1 week at °C PuX2-x Decomposition to form other ratios Direct from elements Structures All PuX are fcc All Pu2X3 are bcc or orthorhombic PuX2-x are tetragonal with a PuS2 monoclinic PuTe3 pseudo-tetragonal

122
Pu S, Se, Te Properties Metallic luster PuS: Au color PuSe: Cu color
PuTe: Black Nonmagnetic Semiconductors f-d hybridization

123
Alkali metal oxoplutonates
Formed from PuO2 and alkali metal oxides, hydroxides, peroxides, or carbonates Variation in atmosphere O2, inert gas, vacuum Pu(IV) Li8PuO6 Pu(V) Li7PuO6, Li3PuO4, Na3PuO4 Oxidizing atmosphere Pu(VI) M6PuO6, M4PuO5 (Li, Na) M2PuO4 (K, Rb, Cs) From oxides in oxygen atmosphere Pu(VII) M5PuO4 (Li, Na) M3PuO5 (Rb, Cs)
 Li8PuO6. Pu(V) Li7PuO6, Li3PuO4, Na3PuO4. Oxidizing atmosphere. Pu(VI) M6PuO6, M4PuO5 (Li, Na) M2PuO4 (K, Rb, Cs) From oxides in oxygen atmosphere. Pu(VII) M5PuO4 (Li, Na) M3PuO5 (Rb, Cs)")
124
Group II Pu oxides Pu(III) BaPu2O4 BaO, Pu, and PuO2 in H2
Formation of PuHx from Pu metal Atmosphere switched to inert Pu(IV) Sr and Ba compounds only MPuO3 Pu(V) Ba3PuO5.5 From Ba3PuO6, PuO2, and BaO Oxidation state of compound is uncertain Pu(VI) MPuO4, M3PuO6 (Ca, Sr, and Ba) Ba2MPuO6 (Sr, Mn, Pb, Mg, Ca)
 Sr and Ba compounds only. MPuO3. Pu(V) Ba3PuO5.5. From Ba3PuO6, PuO2, and BaO. Oxidation state of compound is uncertain. Pu(VI) MPuO4, M3PuO6 (Ca, Sr, and Ba) Ba2MPuO6 (Sr, Mn, Pb, Mg, Ca)")
125
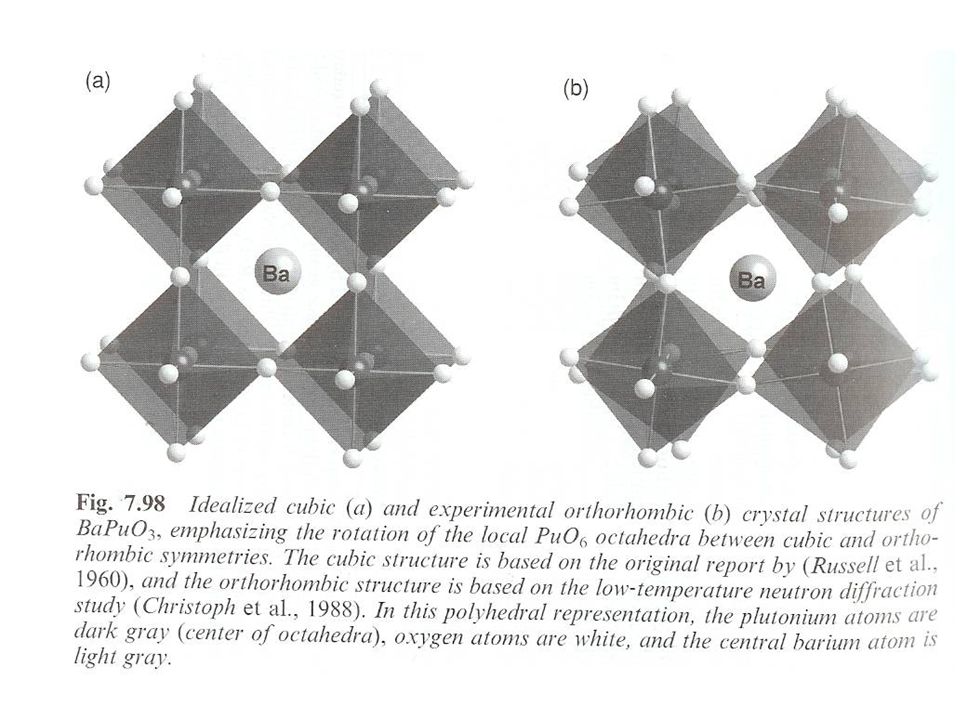
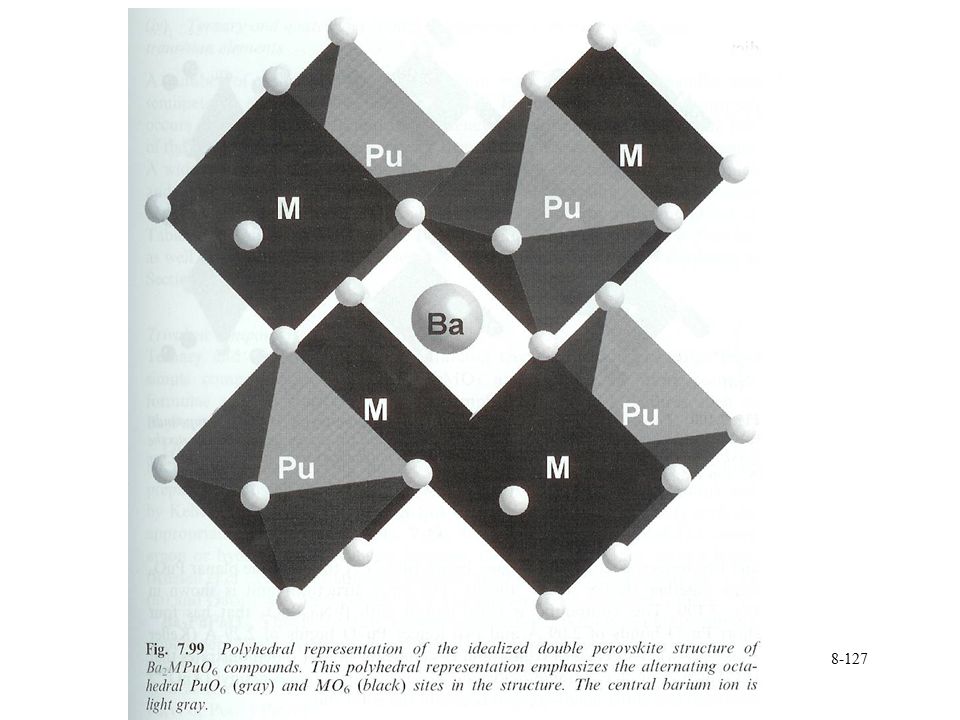
Structures Perovskites CaTiO3 structure (ABO3)
Pu(IV, VI, or VII) in octahedral PuO6n- Cubic lattice BO6 octahedra with A cations at center unit cell Double perovskites (Ba,Sr)3PuO6 and Ba(Mg,Ca,Sr,Mn,Zn)PuO6 M and Pu(VI) occupy alternating octahedral sites in cubic unit cell
 in octahedral PuO6n- Cubic lattice. BO6 octahedra with A cations at center unit cell. Double perovskites. (Ba,Sr)3PuO6 and Ba(Mg,Ca,Sr,Mn,Zn)PuO6. M and Pu(VI) occupy alternating octahedral sites in cubic unit cell.")
129
Pu-Ln oxides PuO2 mixed with LnO1.5 Form solid solutions Oxidation of Pu at higher levels of Ln oxides to compensate for anion defects Solid solutions with CeO2 over entire range

130
Ternary oxides of Pu and actinides
Prepared with Th, Pa, U, and Cm ThO2 Solid solutions over entire range Follows Vegard’s Law At 1000 °C Melting points constant up to 25 % wt ThO2, increase linearly with increasing ThO2 At 1650 °C under Ar partial phase separtion C-Pu2O3

131
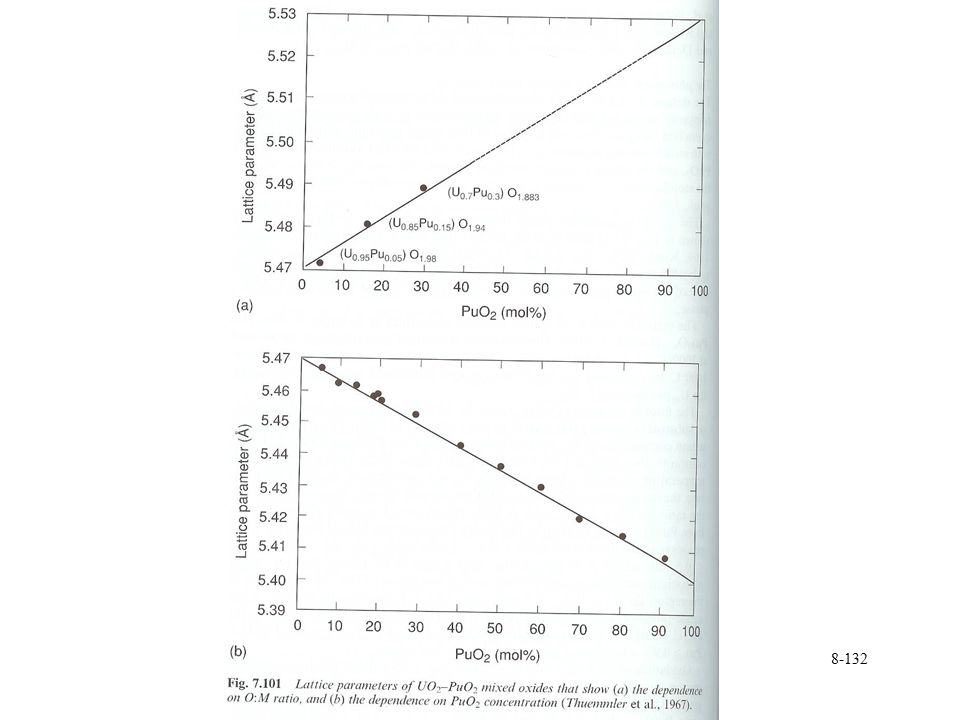
U-Pu-Oxides MOX fuel 2-30 % PuO2 Lattice follows Vegard’s law
Different regions Orthorhombic U3O8 phase Flourite dioxide Deviations from Vegard’s law may be observed from O loss from PuO2 at higher temperature

133
Prepared by precipitation process or co-milling
Properties examined O potential Thermal

134
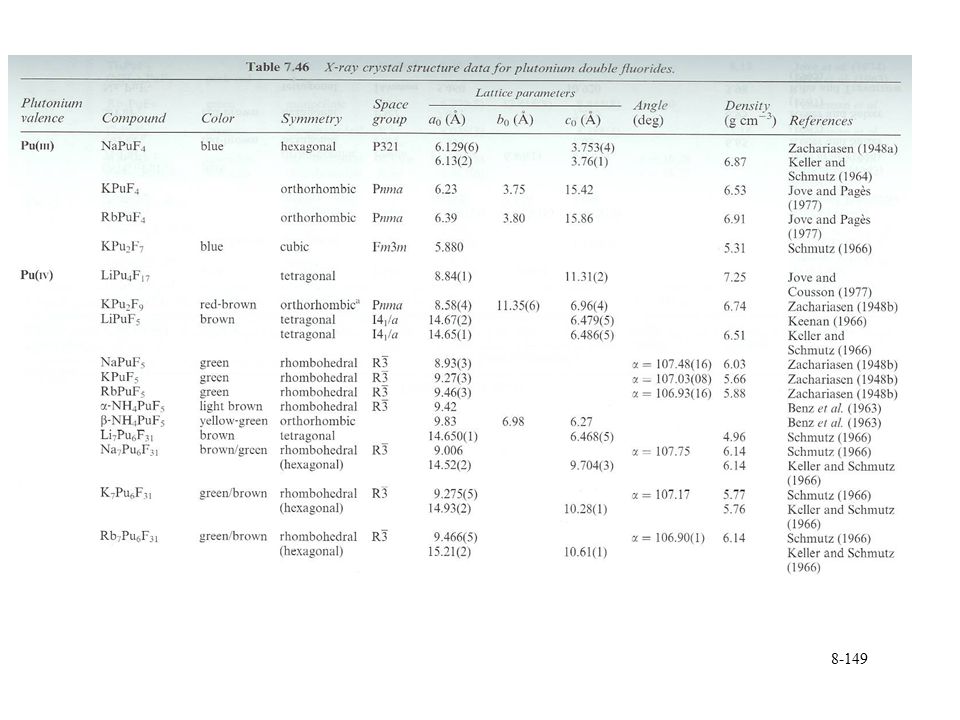
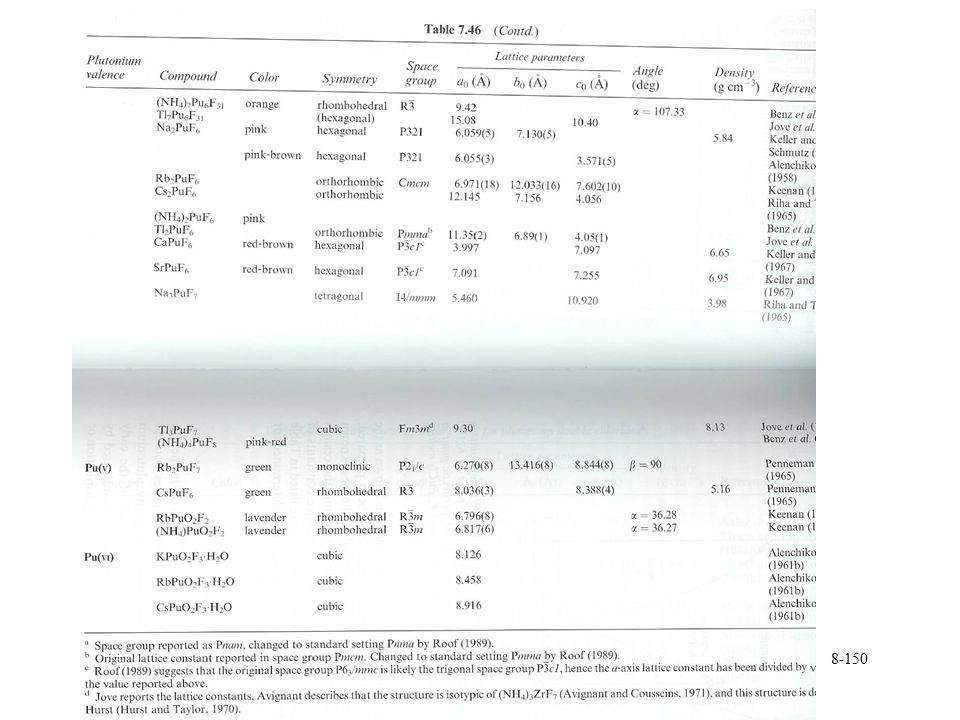
Plutonium halides General formula PuX3
PuF4 stable solid, PuCl4 can be found in gas phase PuF6 gas phase f2 electron configuration Trivalent oxyhalides PuOX Some different oxyfluorides can be formed PuOF3, PuOF4, PuO2F2 A range of fluoride salts with monovalent cation General formula MxPuF4+x x = 1, 2, 3, 4 M7Pu6F31 and MPuF6

135
Pu fluoride preparation
Used in the preparation of Pu metal 2PuO2 + H2 +6 HF 2 PuF3 + 4 H2O at 600 °C Pu2(C2O4)3 + 6 HF2 PuF3 + 3 CO + 3 CO2 + 3 H2O at 600 °C At lower temperature (RT to 150 °C) Pu(OH)2F2 or Pu(OH)F3 forms PuF3 from HF and H2 PuF4 from HF and O2 Other compounds can replace oxalates (nitrates, peroxides) Stronger oxidizing conditions can generate PuF6 PuO2 + 3 F2 PuF6 + O2 at 300 °C PuF4 + F2 PuF6 at 300 °C
3 + 6 HF2 PuF3 + 3 CO + 3 CO2 + 3 H2O at 600 °C. At lower temperature (RT to 150 °C) Pu(OH)2F2 or Pu(OH)F3 forms. PuF3 from HF and H2. PuF4 from HF and O2. Other compounds can replace oxalates (nitrates, peroxides) Stronger oxidizing conditions can generate PuF6. PuO2 + 3 F2 PuF6 + O2 at 300 °C. PuF4 + F2 PuF6 at 300 °C.")
136
Pu fluoride preparation
Insoluble in water Prepared from addition of HF to Pu(III) solution Reduce Pu(IV) with hydroxylamine (NH2OH) or SO2 Purple crystals PuF3.0.40H2O Anhydrous PuF3 formed by heating in HF gas at °C heating starting material in H2 from 150 to 600 °C, then HF at °C Heating PuF4 in H2 from 600 °C
 solution. Reduce Pu(IV) with hydroxylamine (NH2OH) or SO2. Purple crystals. PuF3.0.40H2O. Anhydrous PuF3 formed by heating in HF gas at °C. heating starting material in H2 from 150 to 600 °C, then HF at °C. Heating PuF4 in H2 from 600 °C.")
137
Pu fluoride preparation
In soluble in H2O From the addition of HF to Pu(IV) solution Pale pink PuF4.2.5H2O Soluble in nitric acid solutions that form fluoride species Zr, Fe, Al, BO33- Heating under vacuum yields trifluoride Formation of PuO2 from reaction with water PuF4+2H2OPuO2+4HF Reaction of oxide with fluoride 3PuF4+2PuO24PuF3+O2 Net: 4PuF4+2H2O4PuF3+4HF+O2 High vacuum and temperature favors PuF3 formation Anhydrous forms in stream of HF gas
 solution. Pale pink PuF4.2.5H2O. Soluble in nitric acid solutions that form fluoride species. Zr, Fe, Al, BO33- Heating under vacuum yields trifluoride. Formation of PuO2 from reaction with water. PuF4+2H2OPuO2+4HF. Reaction of oxide with fluoride. 3PuF4+2PuO24PuF3+O2. Net: 4PuF4+2H2O4PuF3+4HF+O2. High vacuum and temperature favors PuF3 formation. Anhydrous forms in stream of HF gas.")
138
Pu fluoride preparation
Formation from reaction of F2 and PuF4 Fast rate of formation above 300 °C Reaction rate Log(rate/mg PuF4 cm-2hr-1= /T) Faster reaction at 0.8 F2 partial pressure Condensation of product near formation Liquid nitrogen in copper condenser near PuF4 Can be handled in glass
 Faster reaction at 0.8 F2 partial pressure. Condensation of product near formation. Liquid nitrogen in copper condenser near PuF4. Can be handled in glass.")
139
Pu fluoride structures
Each Pu surrounded by 9 F

140
Pu fluoride structures
Isostructural with An and Ln tetraflourides Pu surrounded by 8 F Distorted square antiprism PuF6 Gas phase Oh symmetry

141
Pu fluoride properties
Melting point: 1425 °C Boiling point: decomposes at 2000 °C PuF4 Melting point: 1037°C PuF6 Melting point: 52°C Boiling point: 62°C ΔsublH°=48.65 kJ/mol, ΔfH°= kJ/mol IR active in gas phase, bending and stretching modes Isotopic shifts reported for 239 and 242

143
PuF6 Equilibrium constant measured for PuF6PuF4+F2 ΔG=2.55E4+5.27T At 275 °C, ΔG=28.36 kJ/mol ΔS=-5.44 J/K mol ΔH=25.48 kJ/mol

144
Pu halides PuF6 decomposition Alpha decay and temperature
Exact mechanism unknown Stored in gas under reduced pressure Higher halide preparation PuCl3 from hydrochlorination Pu2(C2O4)3.10H2O+6HCl2PuCl3+3CO2+3CO+13H2O Reaction of oxide with phosgene (COCl2) at 500 °C Evaporation of Pu(III) in HCl solution PuCl4 PuCl3+0.5Cl2PuCl4 Gas phase Identified by peaks in gas phase IR
3.10H2O+6HCl2PuCl3+3CO2+3CO+13H2O. Reaction of oxide with phosgene (COCl2) at 500 °C. Evaporation of Pu(III) in HCl solution. PuCl4. PuCl3+0.5Cl2PuCl4. Gas phase. Identified by peaks in gas phase IR.")
145
Pu halides PuBr3 Combination of elements HBr with PuHx
Pu(III) oxalate with HBr, °C PuI3 Pu metal with HI at 400 °C 2Pu+3HgI2 2PuI3+3Hg Structures (PuCl3) 9 Cl for each Pu Tricapped trigonal prism
 oxalate with HBr, °C. PuI3. Pu metal with HI at 400 °C. 2Pu+3HgI2 2PuI3+3Hg. Structures (PuCl3) 9 Cl for each Pu. Tricapped trigonal prism.")
146
Pu halides PuBr3 and PuI3 structure Isostructural 6 Pu-Br of 3.08 Å
2 Pu-Br caps of 3.06 Å 8 coordinate Pu Br and I larger than Cl Enhanced electron repulsion Properties PuCl3 free energy of formation determined ΔG= T PuOX from reaction with H2O

147
Pu oxyhalides Only prepared with Pu(III) and Pu(VI)
PuOX for all halides from water reaction with PuX3 Pu(VI) species Excess PuF6 with water and HF PuO2F2 PuO2Cl2 from vacuum evaporation of PuCl6 at room temperature
 species. Excess PuF6 with water and HF. PuO2F2. PuO2Cl2 from vacuum evaporation of PuCl6 at room temperature.")
148
Ternary halogenoplutonates
Pu(III-VI) halides with ammonia, group 1, group 2, and some transition metals Preparation Metal halides and Pu halide dried in solution Metal halides and PuF4 or dioxide heat °C in HF stream PuF4 or dioxide with NH4F heated in closed vessel at °C with repeated treatment PuF6 or PuF4 with group 1 or 2 fluorides
 halides with ammonia, group 1, group 2, and some transition metals. Preparation. Metal halides and Pu halide dried in solution. Metal halides and PuF4 or dioxide heat °C in HF stream. PuF4 or dioxide with NH4F heated in closed vessel at °C with repeated treatment. PuF6 or PuF4 with group 1 or 2 fluorides.")
152
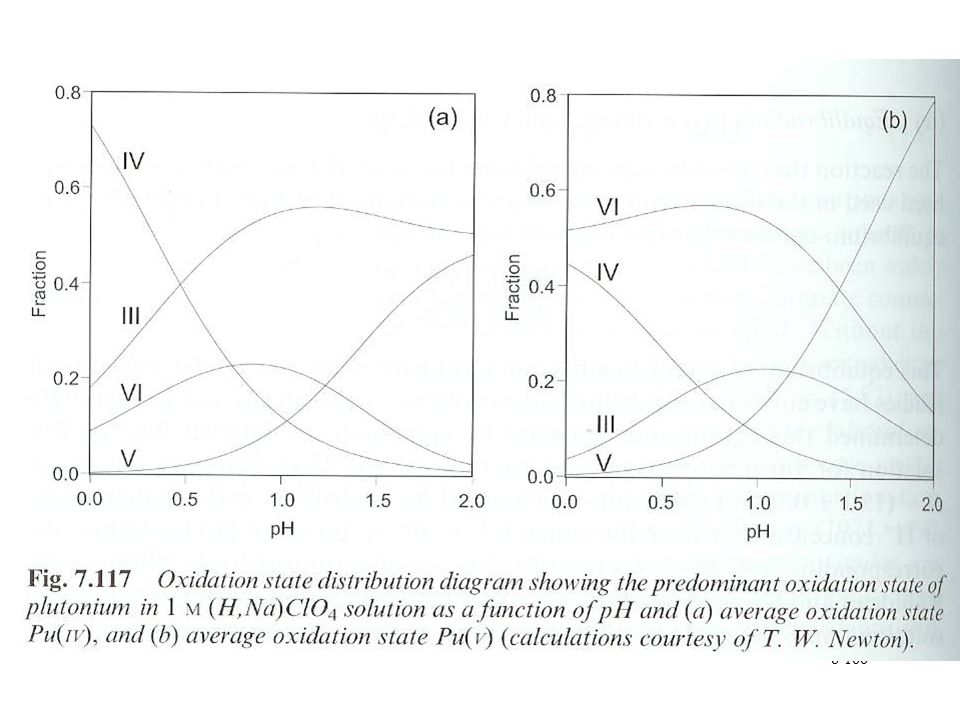
Pu solution chemistry Originally driven by the need to separate and purify Pu Species data in thermodynamic database Complicated solution chemistry Five oxidation states (III to VII) Small energy separations between oxidation states All states can be prepared Pu(III) and (IV) more stable in acidic solutions Pu(V) in near neutral solutions Dilute Pu solutions favored Pu(VI) and (VII) favored in basic solutions Pu(VII) stable only in highly basic solutions and strong oxidizing conditions Some evidence of Pu(VIII)
 Small energy separations between oxidation states. All states can be prepared. Pu(III) and (IV) more stable in acidic solutions. Pu(V) in near neutral solutions. Dilute Pu solutions favored. Pu(VI) and (VII) favored in basic solutions. Pu(VII) stable only in highly basic solutions and strong oxidizing conditions. Some evidence of Pu(VIII)")
153
Pu solution chemistry Pu3+ and Pu4+ simple hydrates free species
Plutonyl oxo species for Pu(V) and Pu(VI) Pu(V) effective charge 2.2 Pu(VI) effective charge 3.2 PuO4- Redox chemistry instrumental in identifying species
 and Pu(VI) Pu(V) effective charge 2.2. Pu(VI) effective charge 3.2. PuO4- Redox chemistry instrumental in identifying species.")
154
Pu solution chemistry Coordination number varies
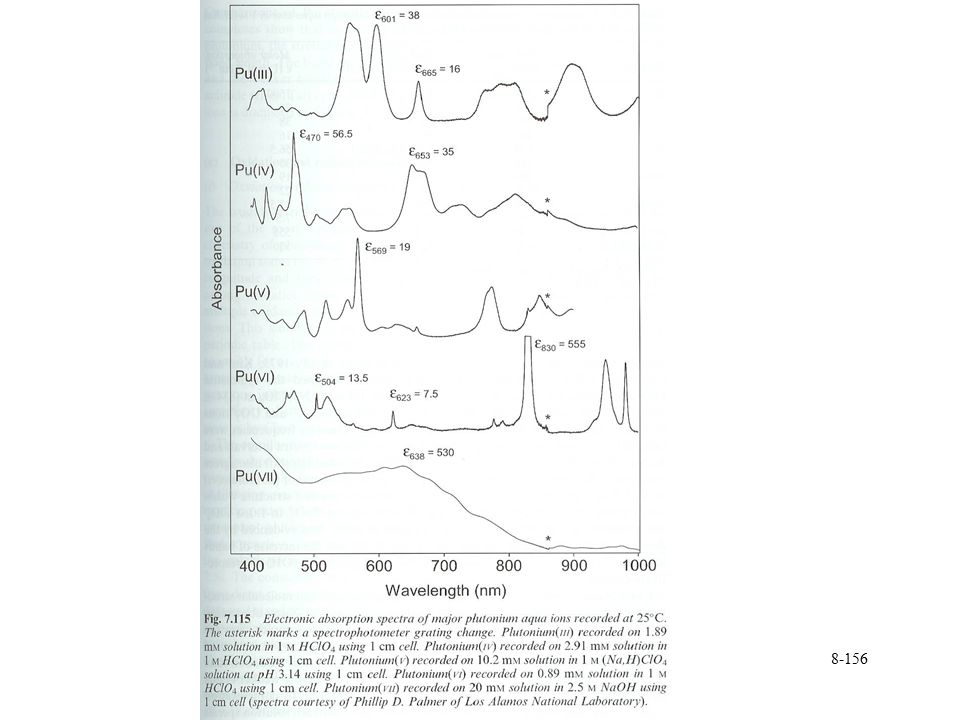
Large values, 8 to 10 for water coordination Spectroscopic properties A few sharp bands 5f-5f transitions More intense than 4f of lanthanides Relativistic effects accentuate spin-orbit coupling Transitions observed spectroscopically Forbidden transitions Sharp but not very intense Pu absorption bands in visible and near IR region Characteristic for each oxidation state

163
Pu solution chemistry Other spectroscopic methods employed in Pu analysis Photoacoustic spectroscopy Thermal lensing Vibrational spectroscopy Oxo species Asymmetric stretch cm-1 962 cm-1 in perchloric acid Linear arrangement of oxygen Raman shifts observed Sensitive to complexation Changes by 40 cm-1

164
Pu solution chemistry Redox chemistry
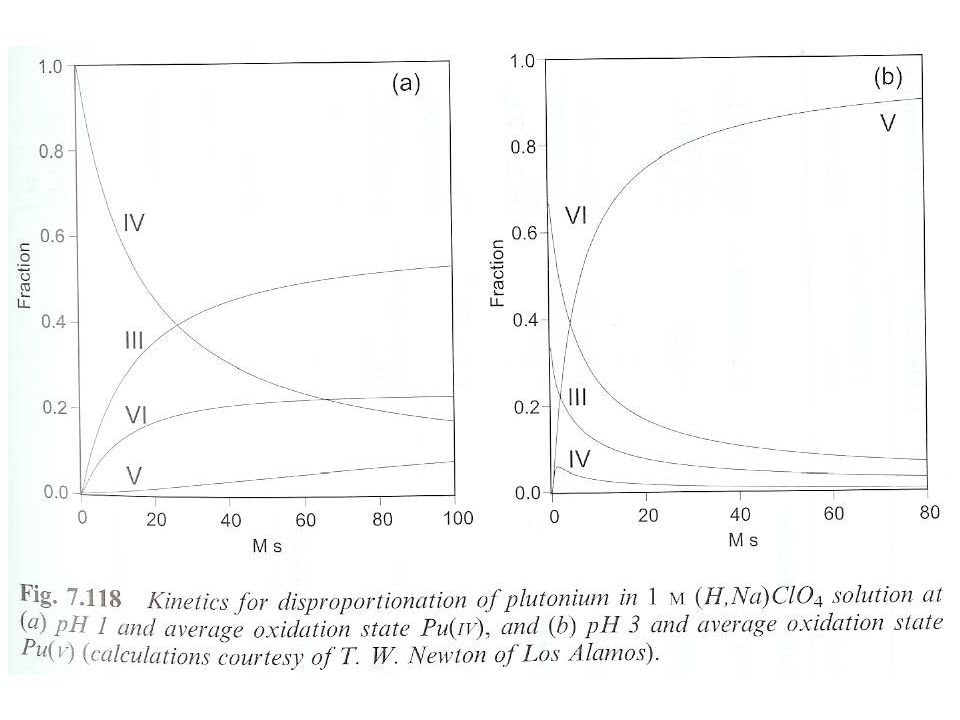
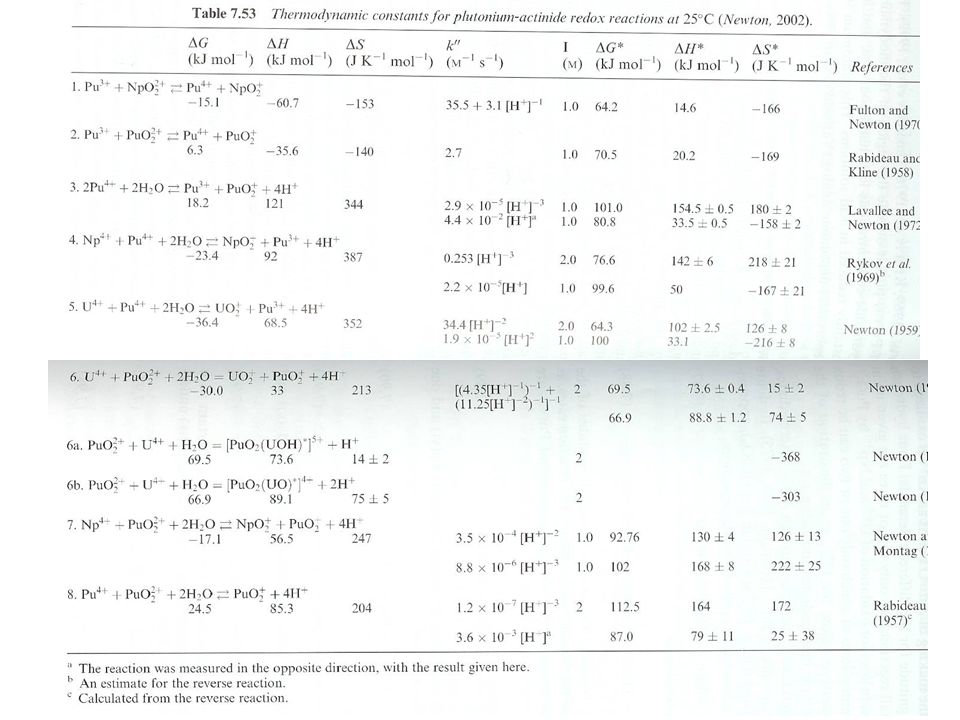
Potentials close to 1 V for 4 common states Kinetics permit coexistance of oxidation states Pu(IV) and Pu(V) tend toward disproportionation 3Pu4++2H2O2Pu3++PuO22++4H+ K= at 1.0 M I 3PuO2++4H+Pu3++2PuO22++2H2O Pu concentration Ionic strength pH Kinetics for disproportionation based on time and Pu concentration Moles seconds (M s) Some redox couples are quasi- or irreversible Breaking or forming oxo bonds i.e., Pu(V)/Pu(III), Pu(VI)/Pu(III) Equilibrium between redox states K=Pu(III)Pu(VI)/Pu(IV)Pu(V) K=13.1, corrected for hydrolysis
 and Pu(V) tend toward disproportionation. 3Pu4++2H2O2Pu3++PuO22++4H+ K= at 1.0 M I. 3PuO2++4H+Pu3++2PuO22++2H2O. Pu concentration. Ionic strength. pH. Kinetics for disproportionation based on time and Pu concentration. Moles seconds (M s) Some redox couples are quasi- or irreversible. Breaking or forming oxo bonds. i.e., Pu(V)/Pu(III), Pu(VI)/Pu(III) Equilibrium between redox states. K=Pu(III)Pu(VI)/Pu(IV)Pu(V) K=13.1, corrected for hydrolysis.")
168
Pu solution chemistry Preparation of pure oxidation states Pu(III)
Generally below pH 4 Dissolve a-Pu metal in 6 M HCl Reduction of higher oxidation state with Hg or Pt cathode 0.75 V vs NHE Hydroxylamine or hydrazine as reductant Pu(IV) Electrochemical oxidation of Pu(III) at 1.2 V Thermodynamically favors Pu(VI), but slow kinetics due to oxo formation Pu(V) Electrochemical reduction of Pu(VI) at pH 3 at 0.54 V (vs SCE) Near neutral in 1 micromole/L Pu(V) Pu(VI) Treatment of lower oxidation states with hot HClO4 Ozone treatment Pu(VII) Oxidation in alkaline solutions Hexavalent Pu with ozone, anodic oxidation
 Electrochemical oxidation of Pu(III) at 1.2 V. Thermodynamically favors Pu(VI), but slow kinetics due to oxo formation. Pu(V) Electrochemical reduction of Pu(VI) at pH 3 at 0.54 V (vs SCE) Near neutral in 1 micromole/L Pu(V) Pu(VI) Treatment of lower oxidation states with hot HClO4. Ozone treatment. Pu(VII) Oxidation in alkaline solutions. Hexavalent Pu with ozone, anodic oxidation.")
169
Pu solution chemistry Pu(VI) oxo oxygen exchange with water
18O enriched water exchange need to maintain hexavalent oxidation state Exchange rate increases with lower oxidation state Exchange half life = 4.55E4 hr at 23 °C Two reaction paths Reaction of water with Pu(VI) Breaking of P=O bonds by alpha decay Faster exchange rate measured with 238Pu Pu redox by actinides Similar to disproprotionation Rates can be assessed against redox potentials Pu4+ reduction by different actinides shows different rates Accompanied by oxidation of An4+ with yl bond formation Reduction of Pu(VI) by tetravalent actinides proceeds over pentavalent state Reactions show hydrogen ion dependency
 Breaking of P=O bonds by alpha decay. Faster exchange rate measured with 238Pu. Pu redox by actinides. Similar to disproprotionation. Rates can be assessed against redox potentials. Pu4+ reduction by different actinides shows different rates. Accompanied by oxidation of An4+ with yl bond formation. Reduction of Pu(VI) by tetravalent actinides proceeds over pentavalent state. Reactions show hydrogen ion dependency.")
171
Pu solution chemistry Pu reduction by other metal ions and ligands
Rates are generally dependent upon proton and ligand concentration Humic acid, oxalic acid, ascorbic acid Poor inorganic complexants can oxidize Pu Bromate, iodate, dichromate Reactions with single electron reductants tend to be rapid Reduction by Fe2+ Complexation with ligands in solution impacts redox Different rates in carbonate media compared to perchlorate Mono or dinitrate formation can effect redox Pu(IV) formation or reaction with pentavalent metal ions proceeds faster in nitrate than perchlorate Oxidation of Pu(IV) by Ce(IV) or Np(VI) slower in nitrate Pu(VI) reduction can be complicated by disproportionation Hydroxylamine (NH2OH), nitrous acid, and hydrazine (N2H4) Used in PUREX for Pu redox control Pu(III) oxidized 2Pu3++3H++NO3-2Pu4++HNO2+H2O Re-oxidation adds nitrous acid to the system which can initiate an autocatalytic reaction
 formation or reaction with pentavalent metal ions proceeds faster in nitrate than perchlorate. Oxidation of Pu(IV) by Ce(IV) or Np(VI) slower in nitrate. Pu(VI) reduction can be complicated by disproportionation. Hydroxylamine (NH2OH), nitrous acid, and hydrazine (N2H4) Used in PUREX for Pu redox control. Pu(III) oxidized. 2Pu3++3H++NO3-2Pu4++HNO2+H2O. Re-oxidation adds nitrous acid to the system which can initiate an autocatalytic reaction.")
172
Pu 2 phase redox system Notes:
The system is a complex two phase system with major reagents including Pu(IV), Pu(III), HAN, nitric acid, and hydrazine. The key is that we do not build up/accelerate the build up of nitrous acid that can trigger an autocatalytic HAN reaction. If the concentration of HAN used in the system is low there is no safety issue. However, concentration of HAN has led to numerous accidents 1/Main issue reduction of Pu and explain that HAN is ONLY in the aqueous phase. The reaction occur in the aqueous phase and/or at the interface. 2/ Reduction of Pu by Hydrazine 3/ Talk about the iron - Fe(II) re-oxidation produces nitrous. (just to be mentioned) 4/ Re-oxidation of Plutonium produces nitrous acid 5/ Nitrous acid partition is much greater into the organic (need an experiment to know – debate with CEA experiment). Biddle and Miles –Rate of Reaction of Nitrous Acid with Hydrazine and with Sulphamic Acid- measured a distribution coefficient of 12.5 at 1.72M nitric acid.
, Pu(III), HAN, nitric acid, and hydrazine. The key is that we do not build up/accelerate the build up of nitrous acid that can trigger an autocatalytic HAN reaction. If the concentration of HAN used in the system is low there is no safety issue. However, concentration of HAN has led to numerous accidents. 1/Main issue reduction of Pu and explain that HAN is ONLY in the aqueous phase. The reaction occur in the aqueous phase and/or at the interface. 2/ Reduction of Pu by Hydrazine. 3/ Talk about the iron - Fe(II) re-oxidation produces nitrous. (just to be mentioned) 4/ Re-oxidation of Plutonium produces nitrous acid. 5/ Nitrous acid partition is much greater into the organic (need an experiment to know – debate with CEA experiment). Biddle and Miles –Rate of Reaction of Nitrous Acid with Hydrazine and with Sulphamic Acid- measured a distribution coefficient of 12.5 at 1.72M nitric acid.")
173
Pu aqueous chemistry Reduction of Pu(IV) to Pu(III) by HAN is fast
Two Reactions are possible: Preferred Reaction depends on the ratio R Comments: It is more unlikely to have reaction 1 occured Hydroxylammonium ion has been considered as the reagent that reduces plutonium. That is not restrictive to our study as the ionization reaction of HAN in nitric acid media is fast. Nitric Acid in solution is dissociated as: H+ and NO3- (cf slide “Ionization Reaction of HAN”) The 1:2 (HAN:Pu) is reaction 1 The 1:1 (HAN:Pu) is reaction 2 Even if R is always <1, our model includes the possibility to go above 1 and also have oscillations or split. References: [1] Kinetic , Mechanism, and Stoicheiometry of the Oxidation of Hydroxylamine by Nitric Acid, G. STEDMAN, J.R. PEMBRIDGE, Journal of Chemical Society, 1657 (1979). [2] Kinetic and product Studies on the Decomposition of Hydroxylamine in Nitric Acid, R.J. GOWLAND, G. STEDMAN, Journal of Inorganic Nuclear Chemisty, 43, (1981).
 The 1:2 (HAN:Pu) is reaction 1. The 1:1 (HAN:Pu) is reaction 2. Even if R is always <1, our model includes the possibility to go above 1 and also have oscillations or split. References: [1] Kinetic , Mechanism, and Stoicheiometry of the Oxidation of Hydroxylamine by Nitric Acid, G. STEDMAN, J.R. PEMBRIDGE, Journal of Chemical Society, 1657 (1979). [2] Kinetic and product Studies on the Decomposition of Hydroxylamine in Nitric Acid, R.J. GOWLAND, G. STEDMAN, Journal of Inorganic Nuclear Chemisty, 43, (1981).")
174
Pu aqueous chemistry kinetic of the reaction derived using the steady state approximation applied to NH2O Reoxidation of Pu rate control With Reference: [3] A Kinetic Study of the Reaction of Plutonium(IV) with Hydroxylamine, G.S. BARNEY, J. Inorg. Nucl. Chem., 38, (1976).
 with Hydroxylamine, G.S. BARNEY, J. Inorg. Nucl. Chem., 38, (1976).")
175
Pu aqueous chemistry In addition to the scavenging of nitrous acid, hydrazine also may reduce Pu(IV) to Pu(III) Excess of Pu Excess of hydrazine Net Autocatalytic reaction may result from Pu redox cycling in HAN/N2H4 system Reference: Kinetics of Reduction of plutonium by Hydrazine. II. Reduction of Pu(IV), KOLTUNOVC, V.S. and ZHURAVLEVA, G.I., , Radiokhimiya, 16 (1), (1974).
, KOLTUNOVC, V.S. and ZHURAVLEVA, G.I., , Radiokhimiya, 16 (1), (1974).")
176
Pu aqueous chemistry Autoradiolysis
Formation of radicals and redox agents Low reaction if concentrations below 1 M With nitrate can form other reactive species (HNO2) Formation of Pu(IV).H2O2 Rate proportional to Pu concentration and dose rate Pu(VI) reduction proceeds over Pu(V) Formation of HNO2 and disproportionation
 Formation of Pu(IV).H2O2. Rate proportional to Pu concentration and dose rate. Pu(VI) reduction proceeds over Pu(V) Formation of HNO2 and disproportionation.")
177
Pu hydrolysis Size and charge
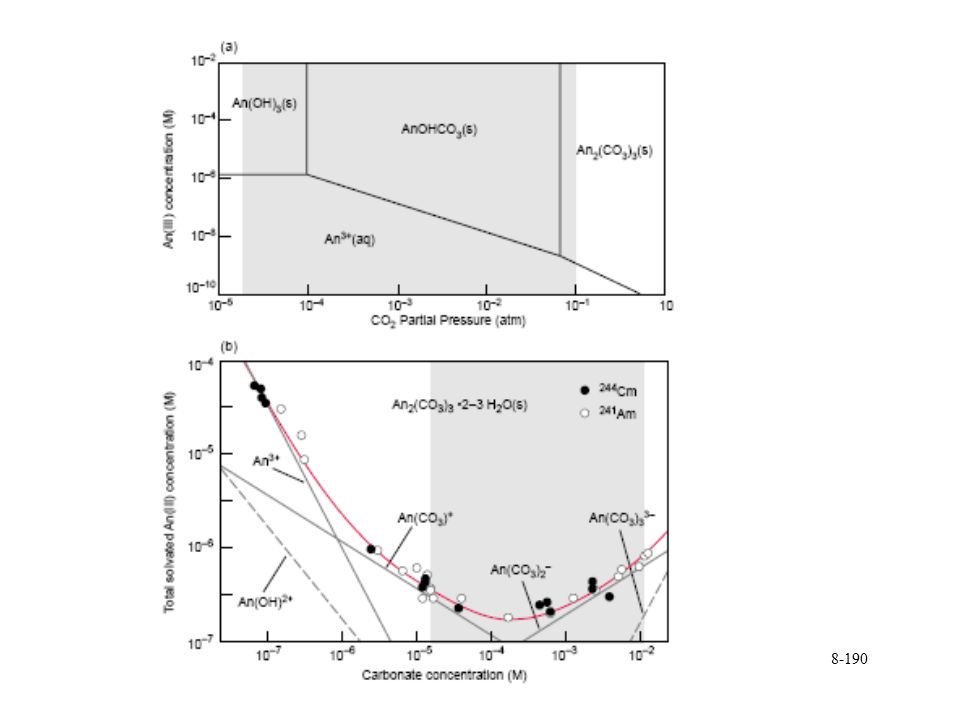
Smaller ions of same charge higher hydrolysis For tetravalents Pu>Np>U>Pa>Th

178
Pu hydrolysis 10 mM

179
Pu(III) 10 mM
 10 mM")
180
Pu(IV) 10 mmol/L
 10 mmol/L")
181
Pu(V) 10 mmol/L
 10 mmol/L")
182
Pu(VI) 10 mmol/L
 10 mmol/L")
183
Pu aqueous chemistry Hydrolysis/colloid formation
In many systems solubility derived Pu(IV) concentrations vary due to colloid formation Colloids are 1- to 1000-nm size particles that remain suspended in solution x-ray diffraction patterns show Pu(IV) colloids are similar to the fcc structure of PuO2 Basis for theory that colloids are tiny crystallites PuO2, May include some water saturated of hydrated surface Prepared by addition of base or water to acidic solutions
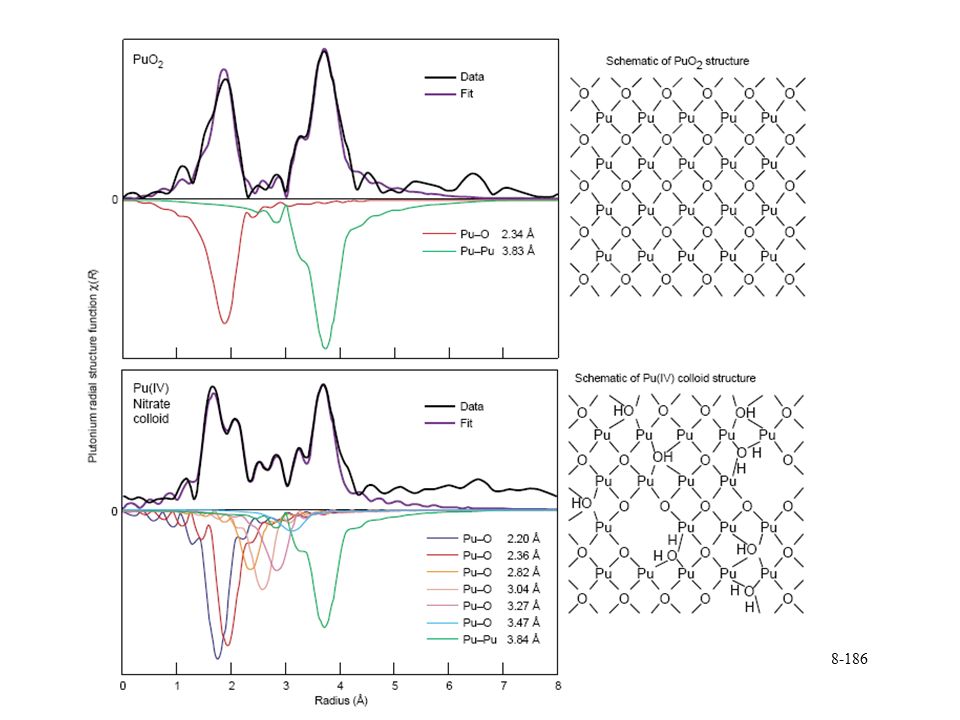
 concentrations vary due to colloid formation. Colloids are 1- to 1000-nm size particles that remain suspended in solution. x-ray diffraction patterns show Pu(IV) colloids are similar to the fcc structure of PuO2. Basis for theory that colloids are tiny crystallites PuO2, May include some water saturated of hydrated surface. Prepared by addition of base or water to acidic solutions.")
184
Pu colloid model

185
Pu aqueous chemistry: colloids
Characterization SANS Long, thin rods 4.7 nm x 190 nm Light scattering Spherical particles 1 nm to 370 nm Laser induced breakdown 12 nm to 25 nm XAFS studies of Pu(IV) colloids demonstrated that average fcc structure is overly simplistic additional chemical forms are present that affect solubility Variations in measured Pu(IV) concentrations may be related to the local structure colloids displays many discrete Pu–O distances 2.25 Å Pu-OH to 3.5 Å amplitude of Pu–Pu is reduced, decrease in number of nearest neighbors four H atoms incorporated into the Pu(IV) colloid structure could result in one Pu vacany. EXAFS reveals that many atoms in the colloid structure are distributed in a non-Gaussian way when several different oxygen containing groups are present O2–,, OH-, and OH2
 colloids. demonstrated that average fcc structure is overly simplistic. additional chemical forms are present that affect solubility. Variations in measured Pu(IV) concentrations may be related to the local structure. colloids displays many discrete Pu–O distances Å Pu-OH to 3.5 Å. amplitude of Pu–Pu is reduced, decrease in number of nearest neighbors. four H atoms incorporated into the Pu(IV) colloid structure could result in one Pu vacany. EXAFS reveals that many atoms in the colloid structure are distributed in a non-Gaussian way when. several different oxygen containing groups are present. O2–,, OH-, and OH2.")
187
Pu aqueous chemistry Complexing ions
General oxidation state trends for complexation constants Pu(IV)>Pu(VI)≈Pu(III)>Pu(V) Oxoanions Pu complexes based on charge and basicity of ligand ClO4-<IO3-<NO3-<SO42-<<CO32-<PO43- 7 to 12 ligands (higher value for Pu(IV) Carbonate Inner and outer sphere complexation with water Outer interaction form chains and layer structures Bidentate with small bite angle Pu(III) carbonate Oxidize rapidly to tetravalent state Complexation values consistent with Am(III) Pu(IV) carbonate Pu(CO3)n4-2n, n from 1 to 5 n increases with pH and carbonate concentration
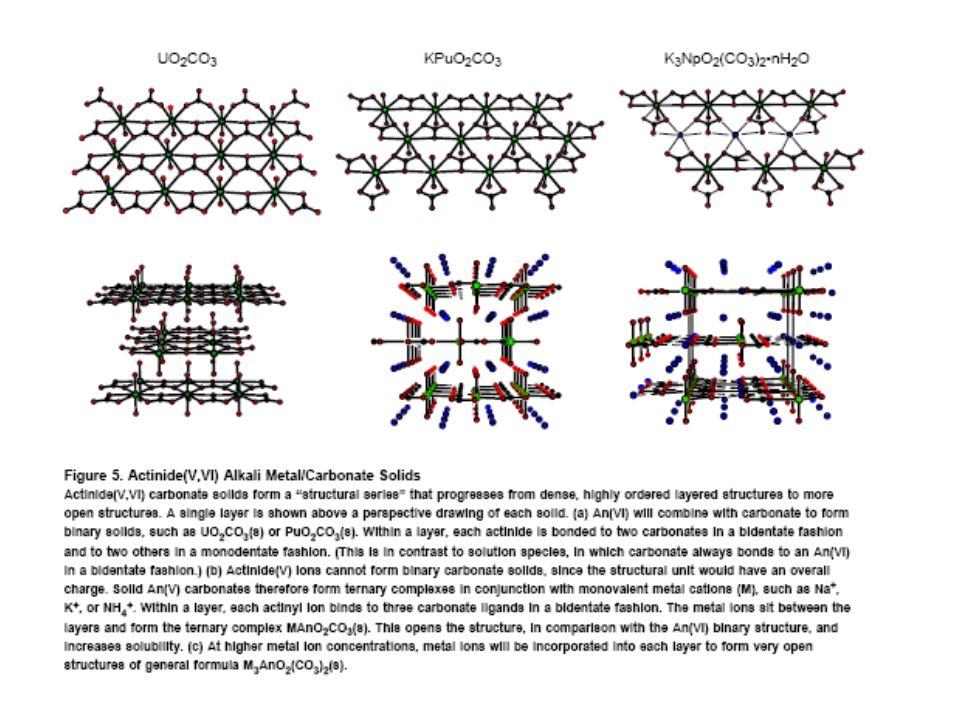
>Pu(VI)≈Pu(III)>Pu(V) Oxoanions. Pu complexes based on charge and basicity of ligand. ClO4-<IO3-<NO3-<SO42-<<CO32-<PO43- 7 to 12 ligands (higher value for Pu(IV) Carbonate. Inner and outer sphere complexation with water. Outer interaction form chains and layer structures. Bidentate with small bite angle. Pu(III) carbonate. Oxidize rapidly to tetravalent state. Complexation values consistent with Am(III) Pu(IV) carbonate. Pu(CO3)n4-2n, n from 1 to 5. n increases with pH and carbonate concentration.")
189
Pu aqueous chemistry Pu(V) carbonates Carbonates to Pu(V) solution
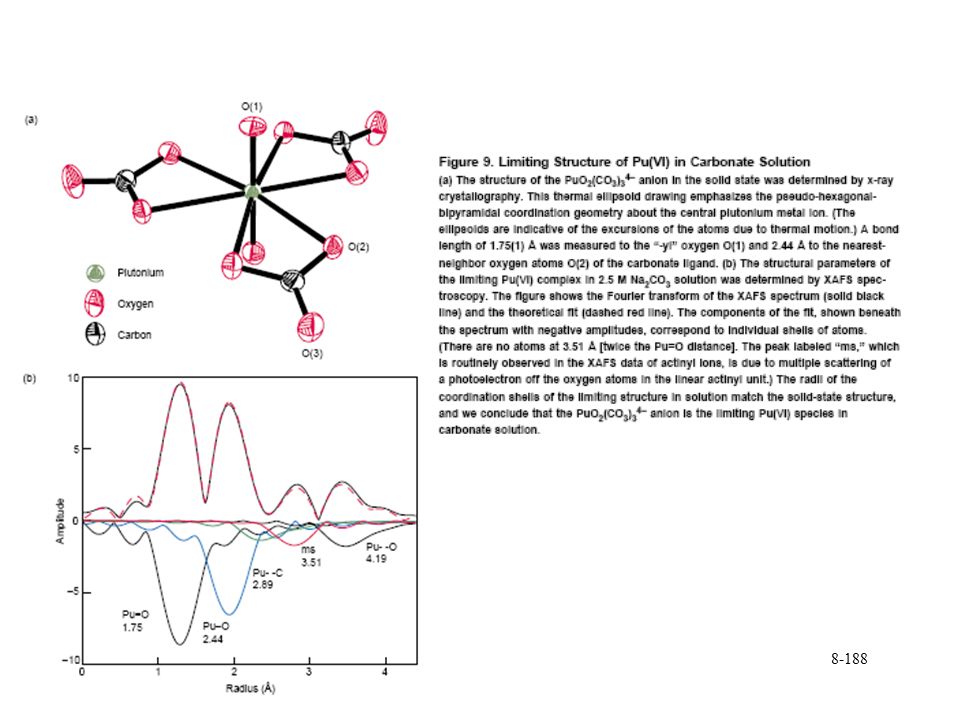
Reduction of Pu(VI) carbonates Mono and triscarbonato species Pu(VI) extension of U(VI) chemistry
 carbonates. Mono and triscarbonato species. Pu(VI) extension of U(VI) chemistry.")
193
Pu solution chemistry Pu nitrates
First Pu complexes and important species in reprocessing and separations Bidentate and planar geometry Similar to carbonates but much weaker ligand 1 or more nitrates in inner sphere Pu(III) species have been prepared but are unstable Pu(IV) species Pu(NO3)n4-n, n=1-6 Tris and pentanitrato complexes not as prevalent Removal of water from coordination sphere with nitrate complexation Pu-O; 2.49 Å for Nitrate, 2.38 Å for H2O Spectrophotometric determination of complexation constants with nitrate and perchlorate Pu(NO3)66- complexes with anion exchange resin For Pu(IV) unclear if penta- or hexanitrato species Evidence suggests hexanitrato species in the presence of resins
 species have been prepared but are unstable. Pu(IV) species. Pu(NO3)n4-n, n=1-6. Tris and pentanitrato complexes not as prevalent. Removal of water from coordination sphere with nitrate complexation. Pu-O; 2.49 Å for Nitrate, 2.38 Å for H2O. Spectrophotometric determination of complexation constants with nitrate and perchlorate. Pu(NO3)66- complexes with anion exchange resin. For Pu(IV) unclear if penta- or hexanitrato species. Evidence suggests hexanitrato species in the presence of resins.")
194
Pu solution chemistry Pu nitrates
Nitrate solids from precipitation from nitric acid solutions Orthorhombic Pu(NO3)4..5H2O M2Pu(NO3)6.2H2O; M=Rb, Cs, NH4+, pyridinium in 8 to 14 M HNO3 Pu-O Å Mixed species TBP complexes, amide nitrates No inner sphere Pu(V) nitrate complexes found Only Pu(VI) mononitrate in solution Solid phase PuO2(NO3)2.xH2O; x=3,6 characterized
4..5H2O. M2Pu(NO3)6.2H2O; M=Rb, Cs, NH4+, pyridinium in 8 to 14 M HNO3. Pu-O Å. Mixed species. TBP complexes, amide nitrates. No inner sphere Pu(V) nitrate complexes found. Only Pu(VI) mononitrate in solution. Solid phase PuO2(NO3)2.xH2O; x=3,6 characterized.")
195
Pu solution chemistry Sulfate Pu(III) Mono and disulfate complexes
Solid K5Pu(SO4)4.8H2O Indicates Pu(SO4)45- in solution Likely Pu(SO4)n3-2n in solution Pu(IV) High affinity for sulfate complexes Mono and bisulfate solution species Solid K4Pu(SO4)4.2H2O hydrated Pu(SO4)2 n=4, 6, 8, 9 Mixed Pu2(OH)2(SO4)3(H2O)4 Should be in basic solution with high sulfate Pu(V) species not well characterized Pu(VI) forms mono- and bisulfate from acidic solutions Examined by optical and IR spectroscopy Solids of M2PuO2(SO4)2
4.8H2O. Indicates Pu(SO4)45- in solution. Likely Pu(SO4)n3-2n in solution. Pu(IV) High affinity for sulfate complexes. Mono and bisulfate solution species. Solid K4Pu(SO4)4.2H2O. hydrated Pu(SO4)2 n=4, 6, 8, 9. Mixed Pu2(OH)2(SO4)3(H2O)4. Should be in basic solution with high sulfate. Pu(V) species not well characterized. Pu(VI) forms mono- and bisulfate from acidic solutions. Examined by optical and IR spectroscopy. Solids of M2PuO2(SO4)2.")
196
Pu solution chemistry Phosphate complexes Low solubility
Range of solid species, difficult characterization Range of protonated phosphates P2O74-, (PO3)nn- Ternary complexes Halides, organics, uranium Pu(III) Not characterized but proposed Pu(H2PO4)n3-n n=1-4 Pu(IV) Wide range of complexes Only Pu(HPO4)2.xH2O examined in solution phase Pu(V) Ammonium monohydratephosphate Pu(V) tetrahydrate species Evidence of PuO2HPO4- Pu(VI) MPuO2PO4.yH2O Solution complexes from Pu(VI) hydroxide and H3PO4
nn- Ternary complexes. Halides, organics, uranium. Pu(III) Not characterized but proposed. Pu(H2PO4)n3-n n=1-4. Pu(IV) Wide range of complexes. Only Pu(HPO4)2.xH2O examined in solution phase. Pu(V) Ammonium monohydratephosphate Pu(V) tetrahydrate species. Evidence of PuO2HPO4- Pu(VI) MPuO2PO4.yH2O. Solution complexes from Pu(VI) hydroxide and H3PO4.")
197
Pu solution chemistry Iodate Pu(IO3)4 precipitate
Not well characterized Prepared by hydrothermal methods Preparation of Pu(VI) diiodate species Mixed Pu(VI) trishydroxide species From Pu(IV) and H5IO6 in hydrothermal reaction, forms (PuO2)2(IO3)(m-OH)3 Pu(V) forms Pu(IV/VI) species Perchlorate No pure solution or solid phases characterized Most likely does not form inner sphere complexes in aqueous solution Oxalates Previously discussed, forms microcrystals Mono and bidentate forms Pu(III) form trivalent oxalates with 10 and 6 hydrates Pu(IV) forms with 2, 4, and 5 oxalates with n waters (n=0,1,2,or 6) Tetra and hexa monovalent M salts Mono hydroxide mixed solid species formed Pu(V) disproportionates Pu(VI)O2 oxalates
 diiodate species. Mixed Pu(VI) trishydroxide species. From Pu(IV) and H5IO6 in hydrothermal reaction, forms (PuO2)2(IO3)(m-OH)3. Pu(V) forms Pu(IV/VI) species. Perchlorate. No pure solution or solid phases characterized. Most likely does not form inner sphere complexes in aqueous solution. Oxalates. Previously discussed, forms microcrystals. Mono and bidentate forms. Pu(III) form trivalent oxalates with 10 and 6 hydrates. Pu(IV) forms with 2, 4, and 5 oxalates with n waters (n=0,1,2,or 6) Tetra and hexa monovalent M salts. Mono hydroxide mixed solid species formed. Pu(V) disproportionates. Pu(VI)O2 oxalates.")
198
Pu solution chemistry Peroxide
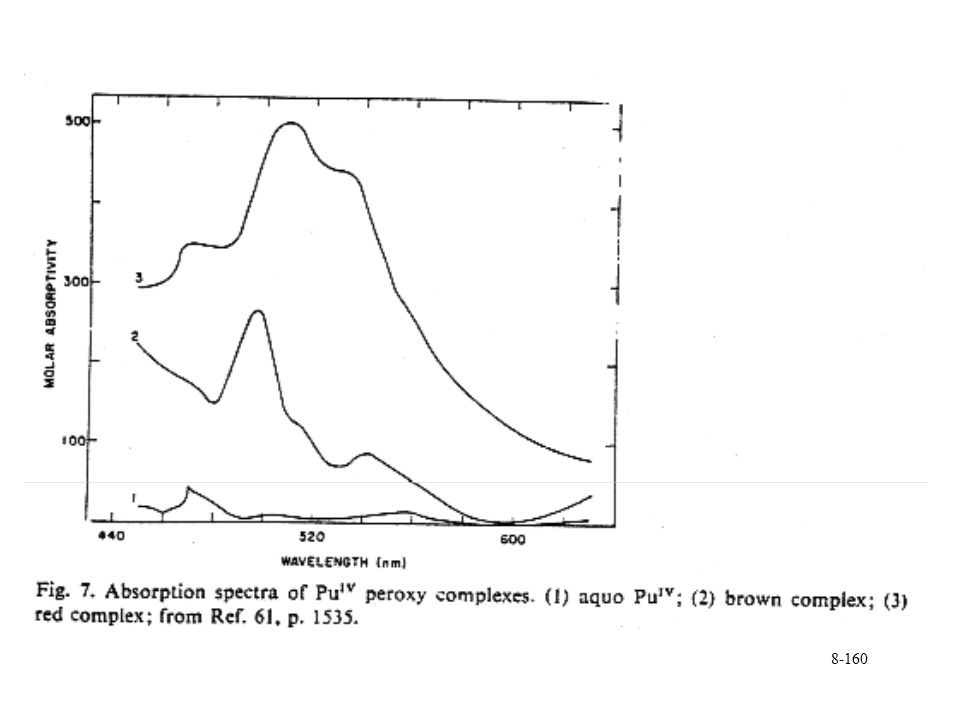
Used to form Pu(IV) from higher oxidation states Further reduction of Pu(IV), mixed oxidation states Pu(IV) peroxide species determined spectroscopically Two different absorbances with spectral change in increasing peroxide No confirmed structure Pu2(m-O2)2(CO3)68- contains doubly bridged Pu-O core Formation of peroxide precipitate that incorporates surrounding anions High acidity and ionic strength In alkaline media, Pu(VI) reduced to Pu(V) with formation of 1:1 complex
 from higher oxidation states. Further reduction of Pu(IV), mixed oxidation states. Pu(IV) peroxide species determined spectroscopically. Two different absorbances with spectral change in increasing peroxide. No confirmed structure. Pu2(m-O2)2(CO3)68- contains doubly bridged Pu-O core. Formation of peroxide precipitate that incorporates surrounding anions. High acidity and ionic strength. In alkaline media, Pu(VI) reduced to Pu(V) with formation of 1:1 complex.")
199
Pu solution chemistry Carboxylate complexes
Single or multiple carboxylate ligands for strong complexes with Pu with typical oxidation state stability trend Tend to stabilize Pu(IV) Pu(III) Oxidation to Pu(IV) at pH > 5 Range of mixed species Degree of protonation (HxEDTA) Mixed hydroxide species Pu(IV) Stabilized by complexation Solution phase at relatively high pH 1:1 Pu to ligand observed (Pu:EDTA, Pu:DTPA) Range of mixed species can be formed EDTA used in the dissolution of Pu(IV) oxide or hydroxide solids Pu(V) complexes to be unstable Oxidation or reduction solution dependent Pu(VI) species observed
 Pu(III) Oxidation to Pu(IV) at pH > 5. Range of mixed species. Degree of protonation (HxEDTA) Mixed hydroxide species. Pu(IV) Stabilized by complexation. Solution phase at relatively high pH. 1:1 Pu to ligand observed (Pu:EDTA, Pu:DTPA) Range of mixed species can be formed. EDTA used in the dissolution of Pu(IV) oxide or hydroxide solids. Pu(V) complexes to be unstable. Oxidation or reduction solution dependent. Pu(VI) species observed.")
200
Pu solution chemistry Halides
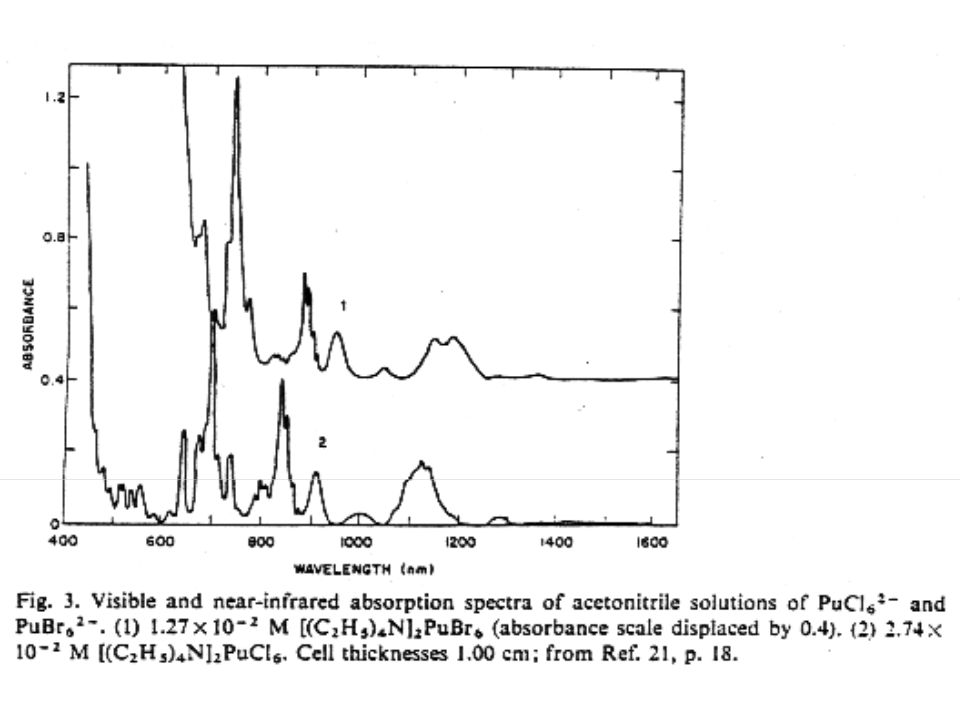
Studies related to Pu separation and metal formation Solid phase double salts discussed Cation-cation complexes Bridging over yl oxygen form plutonyl species Primarily examined for Neptunyl species Observed for UO22+ and PuO2+ 6 M perchlorate solution Formation of CrOPuO4+ cation from oxidation of Pu(IV) with Cr(VI) in dilute HClO4
 with Cr(VI) in dilute HClO4.")
201
Pu non-aqueous chemistry
Very little Pu non-aqueous and organometallic chemistry Limited resources Halides useful starting material Pu halides insoluble in polar organic solvents Formation of solvated complexes PuI3(THF)x from Pu metal with 1,2-diiodoethane in THF Tetrahydrofuran Also forms with pyridine, dimethylsulfoxide Also from the reaction of Pu and I2 Solvent molecules displaced to form anhydrous compounds Single THF NMR environment at room temperature Two structures observed at -90 °C
x from Pu metal with 1,2-diiodoethane in THF. Tetrahydrofuran. Also forms with pyridine, dimethylsulfoxide. Also from the reaction of Pu and I2. Solvent molecules displaced to form anhydrous compounds. Single THF NMR environment at room temperature. Two structures observed at -90 °C.")
202
Pu non-aqueous chemistry
Pu oxidation with Tl or Ag hexafluorophosphate in acetonitrile (CH3CN) Pu(CH3CN)9(PF6)3.CH3CN Pu trivalent cation complex surrounded by PF6- anion 9 coordinate tricapped trigonal prism PuCl4 can be stabilized PuCl4L2or3 from Cs2PuCl6 with amide (RCONR’2) or phosphine oxide (R3PO) Oh based symmetry for L2
 Pu(CH3CN)9(PF6)3.CH3CN. Pu trivalent cation complex surrounded by PF6- anion. 9 coordinate tricapped trigonal prism. PuCl4 can be stabilized. PuCl4L2or3 from Cs2PuCl6 with amide (RCONR’2) or phosphine oxide (R3PO) Oh based symmetry for L2.")
203
Pu non-aqueous chemistry
Amides Uses PuI3(THF)4 starting material 3 NaN(SiMe3)2 yields Pu complex with 3 NaI IR shows asymmetric PuNSi2 stretch at 986 cm-1 Structure based on U and La complexes Alkoxides Reaction of Pu(N(SiMe3)2)3 with 3 2,6-Bu2C6H3OH yields Pu(O-2,6-Bu2C6H3)3 Will coordinate Lewis base (donate electron pair)
4 starting material. 3 NaN(SiMe3)2 yields Pu complex with 3 NaI. IR shows asymmetric PuNSi2 stretch at 986 cm-1. Structure based on U and La complexes. Alkoxides. Reaction of Pu(N(SiMe3)2)3 with 3 2,6-Bu2C6H3OH yields Pu(O-2,6-Bu2C6H3)3. Will coordinate Lewis base (donate electron pair)")
204
Pu non-aqueous chemistry
Borohydrides PuF4 + 2Al(BH4)3Pu(BH4)4+ 2Al(BH4)F2 Separate by condensation of Pu complex in dry ice IR spectroscopy gives pseudo Td 12 coordinate structure Cyclooctatraene (C8H8) complexes [NEt4]2PuCl6 + 2K2C8H8 Pu(C8H8)2+4KCl + 2[NEt4]Cl in THF Slightly soluble in aromatic and chlorinated hydrocarbons D8h symmetry 5f-5f and 5f-6d mixing Covalent bonding, molar absorptivity approaching 1000 L mol-1cm-1
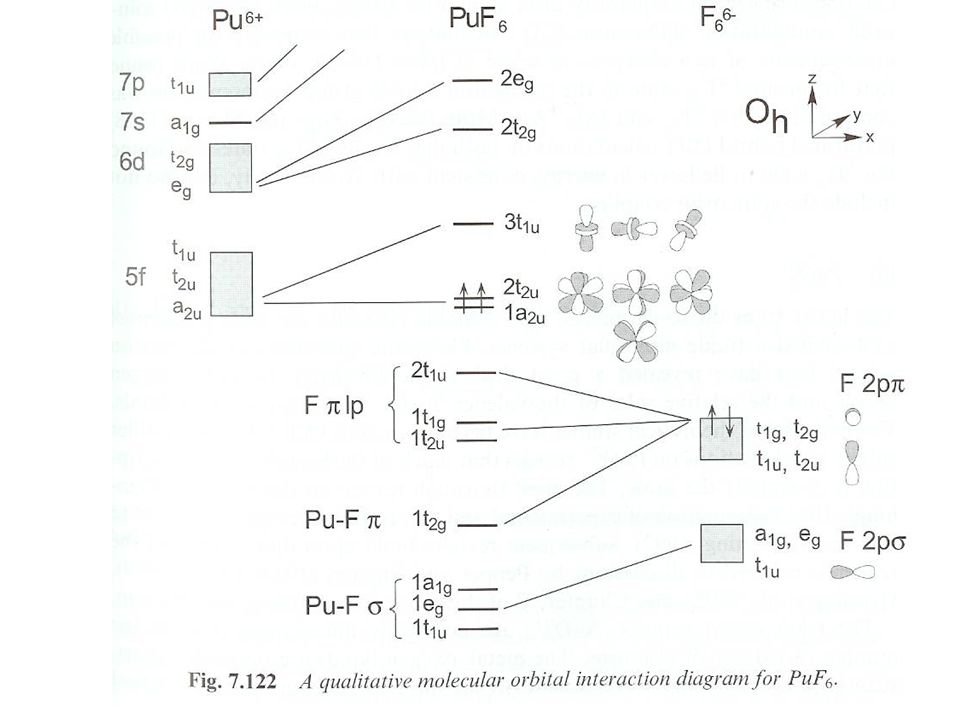
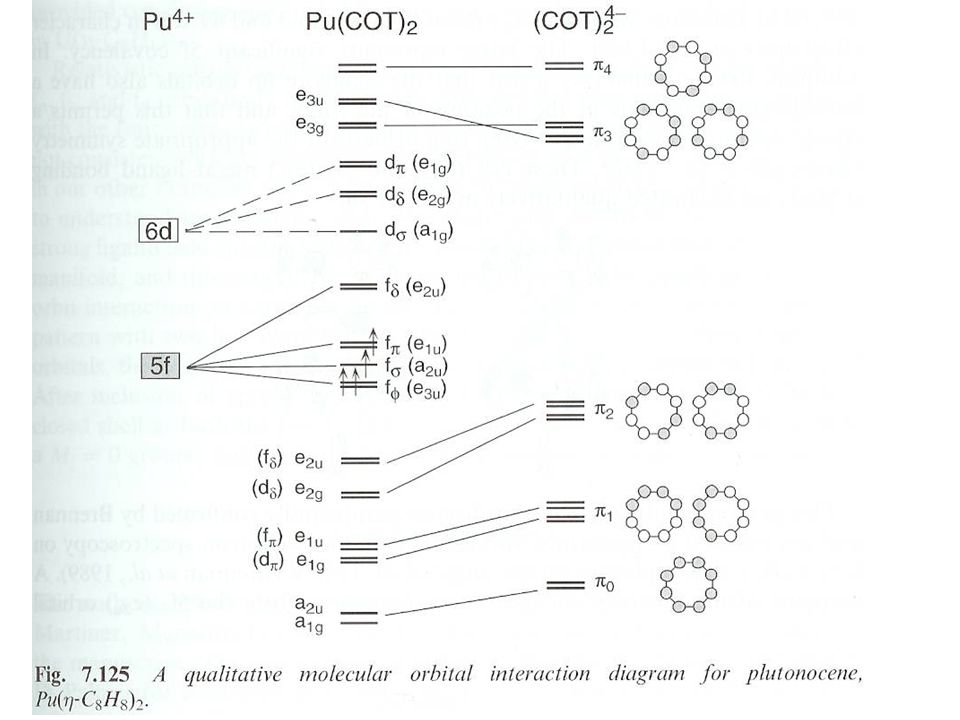
3Pu(BH4)4+ 2Al(BH4)F2. Separate by condensation of Pu complex in dry ice. IR spectroscopy gives pseudo Td. 12 coordinate structure. Cyclooctatraene (C8H8) complexes. [NEt4]2PuCl6 + 2K2C8H8 Pu(C8H8)2+4KCl + 2[NEt4]Cl in THF. Slightly soluble in aromatic and chlorinated hydrocarbons. D8h symmetry. 5f-5f and 5f-6d mixing. Covalent bonding, molar absorptivity approaching 1000 L mol-1cm-1.")
205
Pu non-aqueous chemistry
Cyclopentadienyl (C5H5), Cp PuCl3 with molten (C5H5)2Be trisCp Pu Reactions also possible with Na, Mg, and Li Cp Cs2PuCl6+ 3Tl(C5H5) in acetonitrile Formation of Lewis base species CpPuCl3L2 From PuCl4L2 complex Characterized by IR and Vis spectroscopy
, Cp. PuCl3 with molten (C5H5)2Be. trisCp Pu. Reactions also possible with Na, Mg, and Li Cp. Cs2PuCl6+ 3Tl(C5H5) in acetonitrile. Formation of Lewis base species. CpPuCl3L2. From PuCl4L2 complex. Characterized by IR and Vis spectroscopy.")
206
Pu electronic structure
Ionic and covalent bonding models Ionic non-directional electrostatic bonds Weak and labile in solution Core 5f Covalent bonds are stronger and exhibit stereochemical orientation All electron orbitals need to be considered Evidence of a range of orbital mixing PuF6 Expect ionic bonding Modeling shows this to be inadequate Oh symmetry Sigma and pi bonds t2g interacts with 6d t2u interacts with 5f or 6p and 7p for sigma bonding t1g non-bonding Range of mixing found 3t1u 71% Pu f, 3% Pu p, 26% F p characteristics Spin-orbital coupling splits 5f state Necessary to understand full MO, simple electron filling does not describe orbital 2 electrons in 5f orbital Different arrangements, 7 f states

208
Pu electronic structure
PuO2n+ Linear dioxo Pu oxygen covalency Linear regardless of number of valence 5f electrons D∞h, no Pu oxygen sigma and pi bonds Sigma from 6pz2 and hybrid 5fz3 with 6pz Pi 6d and 5f pi orbitals Valence electrons include non-bonding orbital d and f higher than pi and sigma in energetics 5f add to non bonding orbitals Weak ionic bonds in equatorial plane Spin-orbital calculations shown to lower bond energy

211
Pu electronic structure
Plutonocene 8 C 2 pi orbitals form pi bonds Combine with 6d Pu atomic orbitals 5f form s, p, d, and f orbitals 5fd directed toward C8 rings Bond 49 % f character Demonstrates covalency Other interactions weaker Stronger d interactions Orbital 11% d character

213
Pu electronic structure
Plutonocene Experimental evidence of splitting Need to consider spin-orbital coupling Different occupancy of orbitals

Similar presentations