Download presentation
Presentation is loading. Please wait.

2
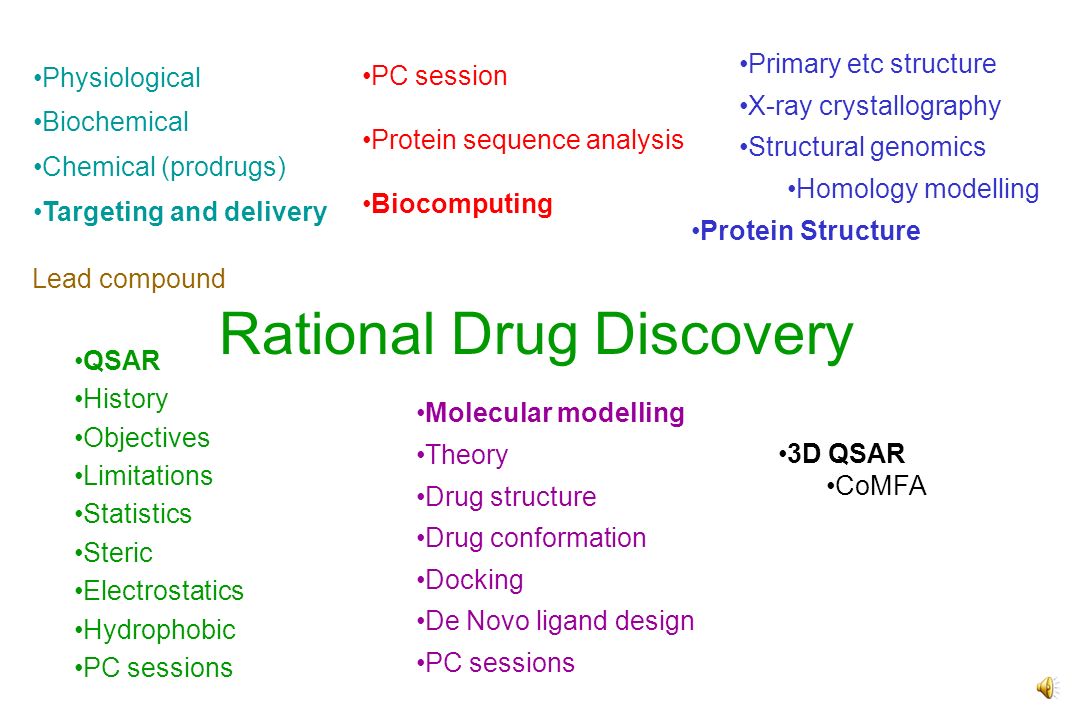
Rational Drug Discovery PC session Protein sequence analysis Biocomputing Primary etc structure X-ray crystallography Structural genomics Homology modelling Protein Structure QSAR History Objectives Limitations Statistics Steric Electrostatics Hydrophobic PC sessions Molecular modelling Theory Drug structure Drug conformation Docking De Novo ligand design PC sessions 3D QSAR CoMFA Lead compound Physiological Biochemical Chemical (prodrugs) Targeting and delivery
 Targeting and delivery")
3
4. Molecular Modelling 4.1. Introduction Molecular modelling can be used to calculate the following that may be useful in drug design Drug structure drugs with a similar structure may show similar activity such drugs can be superimposed - see PC activities structure is required for docking, 3D-QSAR and pharamacophore/receptor mapping - see PC activity Conformation active drugs may share a common conformation - see PC activity inactive drugs may have different conformations Enzyme-drug or receptor drug interactions (geometries and energies) Dipole moments, atomic charges - can be used in QSAR in place of , , logP Electrostatic potentials - molecular with similar electrostatic potentials may have similar activity - see PC activity
 Dipole moments, atomic charges - can be used in QSAR in place of , , logP Electrostatic potentials - molecular with similar electrostatic potentials may have similar activity - see PC activity.")
4
Molecular modelling Docking Geometry optimization/ Global minimum Energy distance Energy Conformation De novo ligand design /automatic docking

5
Bond stretching Torsional rotation Angle bending

6
4.2. Molecular mechanics 4.2.1 Bond stretching. Here we assume the molecules obey classical mechanics a bond is modelled as two balls joined by a spring which obeys Hooke’s law. Energy/kJ mol -1 r/m r eq Bond bond compression stretching k r force constant - a measure of the strength of the bond

7
4.2. Molecular mechanics 4.2.2. Angle bending Here we assume the molecules obey classical mechanics a bond is modelled as two balls joined by a spring which obeys Hooke’s law. Energy/kJ mol -1 /m eq k , angle bending force constant - a measure of ease of bending 20

8
4.2. Molecular mechanics 4.2.3 Torsional energy 90 180 270 360 General enery term is: V N is torsional barrier VNVN 20

9
4.2.4. Non-bonded interactions (1) H-bonds are a result of the electrostatic attraction between + charge on H and - on the O/N atom - see host-guest interaction: C=O……HO-C - + Energy 200 /kJ mol -1 0 -200 Repulsive: + - q i is the charge on atom i r ij is the distance between atoms i, j K is a constant (dielectric constant)
 H-bonds are a result of the electrostatic attraction between + charge on H and - on the O/N atom - see host-guest interaction: C=O……HO-C - + Energy 200 /kJ mol Repulsive: + - q i is the charge on atom i r ij is the distance between atoms i, j K is a constant (dielectric constant).")
10
Sulphonamide binding to carbonic anhydrase Note electrostatic interaction between Zn 2+ (magenta) and N (blue) of 3 His residues and between Zn 2+ and N(Blue) of sulphonamide. (See also and QSAR)
.")
11
4.2.5. Leonard-Jones 12-6 interaction Energy 20 /kJ mol -1 0 -20 Repulsive part - repulsion between electron clouds Attractive part - electrons ‘flicker’ and create a dipole - induces dipole in neighbouring elctron cloud - 2 dipoles are attracted together. This force exists even between 2 inert gas atoms, eg Xe…Xe.

12
4.2.6. Typical molecular mechanics equation The “parameters” k r, k , V N, A, C are determined from experiments on similar compounds The atomic charges, q i, are determined by quantum mechanical calculations, as in nemesis program in PC lab. The accuracy of these calculations depends on how carefully the parameters and charges have been obtained. They may not be valid for very different compounds

13
4.3. Geometry optimisation and drug structure Calculate the energy of the drug Move the atoms to minimize the energy (lowest energy = best structure) Move aided by calculating the forces: F x = -dE/dx etx, where F x is force in x direction on a given atom Repeat until energy is as low as possible - see PC labs Why did some of you get different energies for the same molecules? 30
 Move aided by calculating the forces: F x = -dE/dx etx, where F x is force in x direction on a given atom Repeat until energy is as low as possible - see PC labs Why did some of you get different energies for the same molecules. 30.")
14
4.4. The global minimum problem Energy “geometry” Minimization always finds the nearest minimum, not lowest Start at A, go to B, not C Can be a problem in docking B and C may be related by a conformation change if a side chain rotates it may make a favourable interaction and lower energy C is more likely to be the experimental structure No simple solution to problem - use chemical intuition A BC

15
4.5. Conformation Drugs bind to an enzyme in a particular conformation probably the lowest energy conformation or one close in energy to it Other active compounds probably have to achieve a similar conformation without too large an expenditure of energy Energy variation with conformation studied by torsion driving - see PC labs One torsional angle is fixed, all other bonds, angles, torsional angles optimized until they find lowest energy Fixed torsion is then stepped through say 30° and the optimization repeated Repeat around full 360° Conformation within say 25 kJ mol -1 of global minimum will be accessible Energy 25

16
Digression: contour maps

17
4.6. Conformational energy contour maps 0 360 360 0 A B If energy varies with rotation about 2 torsional angles, plot energy as a contour map Below, left: if compound A and B are both active, the active conformation is…. 0 360 360 0 D C Below, right: If C is active and D is not, active conformation is...

18
4.7. Molecular dynamics (2) One way to find global minimum, C (slide 4.4) is to use MD Here we basically solve Newton’s laws of motion, F=ma Calculate force, F, on each atom of mass, m and let it accelerate, with acceleration a, for a short time of ~0.0001 ps (1 ps = 10 -12 s) Re-evaluate forces and let atoms move again (~10 million moves?) MD is run at a set temperature since the temperature is related to the kinetic energy the temperature can be calculated from the masses and the average velocities. The velocities can be adjusted to keep the temperature constant The motion may mimic the natural vibrations of enzymes, DNA MD, like geometry optimization, is useful for refining X-ray and NMR structures If MD is run at a high temperature, say 800°C, the system may have enough energy to cross high barriers and get to C. It is expensive to run MD for more than 1 ns but many biological structural changes take 1 s - 1 ms so many changes can’t be studied
 One way to find global minimum, C (slide 4.4) is to use MD Here we basically solve Newton’s laws of motion, F=ma Calculate force, F, on each atom of mass, m and let it accelerate, with acceleration a, for a short time of ~ ps (1 ps = s) Re-evaluate forces and let atoms move again (~10 million moves ) MD is run at a set temperature since the temperature is related to the kinetic energy the temperature can be calculated from the masses and the average velocities. The velocities can be adjusted to keep the temperature constant The motion may mimic the natural vibrations of enzymes, DNA MD, like geometry optimization, is useful for refining X-ray and NMR structures If MD is run at a high temperature, say 800°C, the system may have enough energy to cross high barriers and get to C. It is expensive to run MD for more than 1 ns but many biological structural changes take 1 s - 1 ms so many changes can’t be studied.")
19
4.8 Docking Molecular mechanics can be used to find the minimum energy position of a drug within an enzyme Calculated interaction energy may correlate with biological activity Since we may not know how a drug interacts with an enzyme, an approximate starting geometry is usually found using interactive molecular graphics (see PC activity) X-ray crystal structures of analogues or site-directed mutagenesis may be useful in assessing plausible structures (see PC activity) The docked structure may be useful for suggesting improved analogues e.g. regions where the drug may be chemically modified to pick up new interactions and give improved interaction energies

20
4.9. De Novo ligand design There is much activity to write computer programs to analyse space within an active site of an enzyme and automatically design ligands to fit. Such compounds are unlikely to be drugs but could be candidates for high throughput screening (HTS) Such programs may repetitively search a database to find fragments of molecules and automatically test whether they fit Other programs may try to randomly “grow” ligands in active site these programs may test millions of possible structures and so need a quick way to “score” how well ligands fit Instead of using the equations of slides 4.2 they merely count number of hydrogen bonds calculate hyrophobic contact area Such methods are useful for designing appropriate libraries for combinatorial chemistry
 Such programs may repetitively search a database to find fragments of molecules and automatically test whether they fit Other programs may try to randomly grow ligands in active site these programs may test millions of possible structures and so need a quick way to score how well ligands fit Instead of using the equations of slides 4.2 they merely count number of hydrogen bonds calculate hyrophobic contact area Such methods are useful for designing appropriate libraries for combinatorial chemistry.")
21
4.10. Molecular electrostatic potential MEP, (1) MEP at a point P defined as the interaction energy between a molecule and a unit positive charge (i.e. a charge of +1), assuming that the unit charge does not polarize the molecule’s electron distribution It may be used to predict where a molecule will protonate where ia molecule will be attached by a +ve alkylating agent (e.g. DNA - see PC activity) Which part of the molecule will interact with polar/charged regions of enzyme How molecules should align in space +ve MEP regions should align with +ve regions; -ve with -ve Usually displayed as a colour coded surface. It may be calculated by quantum mechanics or by using MEP at P = atoms i q i /r iP where r iP is distance from point P to atom i The atomic charges are calculated using quantum mechanics
 MEP at a point P defined as the interaction energy between a molecule and a unit positive charge (i.e. a charge of +1), assuming that the unit charge does not polarize the molecule’s electron distribution It may be used to predict where a molecule will protonate where ia molecule will be attached by a +ve alkylating agent (e.g. DNA - see PC activity) Which part of the molecule will interact with polar/charged regions of enzyme How molecules should align in space +ve MEP regions should align with +ve regions; -ve with -ve Usually displayed as a colour coded surface. It may be calculated by quantum mechanics or by using MEP at P = atoms i q i /r iP where r iP is distance from point P to atom i The atomic charges are calculated using quantum mechanics.")
22
Beta + gamma Gamma -ve gamma (blue) binds to +ve beta (red/yellow) +ve gamma (yellow/red) binds to -ve beta (blue) 4.10. Molecular electrostatic potential MEP, (2): G beta/gamma 48
: G beta/gamma 48.")
23
4.11. Some limitations of molecular modelling Structural models are sometimes considered to be rigid Solvent/membrane is often ignored So hydrophobic bonding may not be modelled fully and only the L-J 12-6 interaction (between hydrophobic groups) is modelled ionic strength is often ignored molecular mechanics equation is only an approximation (e.g. polarization ignored) quantum effects ignored parameters may be invalid for new types of molecules quantum mechanics quantum effects are often approximated - how else could your charge calculations in PC lab have gone so quickly? Molecular dynamics: time scales may be too short
 is modelled ionic strength is often ignored molecular mechanics equation is only an approximation (e.g. polarization ignored) quantum effects ignored parameters may be invalid for new types of molecules quantum mechanics quantum effects are often approximated - how else could your charge calculations in PC lab have gone so quickly. Molecular dynamics: time scales may be too short.")
24
2.4. Some limitations of QSAR (1) , only valid for substituted benzenes - need to consider diverse structures For new groups: , not available in tables Difficult to deal with inactive compounds (what is activity?) or crude biological data (that may be expressed +, ++, etc) Need to synthesise at least 5 compounds per descriptor Descriptors , , etc are often related to each other so increase in activity due to increase in may actually be due to increase in size Does not address conformation of drug Only gives optimum values of , etc, not structure of new drug. Compounds must be described similarly even if structures are very different (using log P instead of can get round the problem - but then log P has to be measured (but does not as it is found in tables) Can prevent researchers looking at a new series of compounds Often have to use response from a single concentration rather than concentration to achieve set effect - therefore loss of accuracy A single QSAR only addresses one property - may need to consider solubility, stability, absorption, metabolism, transport, safety… QSAR only valid if all compounds in series operate by a common mechanism. This is often not valid. Requires accurate data on weakly active compounds
 , only valid for substituted benzenes - need to consider diverse structures For new groups: , not available in tables Difficult to deal with inactive compounds (what is activity ) or crude biological data (that may be expressed +, ++, etc) Need to synthesise at least 5 compounds per descriptor Descriptors , , etc are often related to each other so increase in activity due to increase in may actually be due to increase in size Does not address conformation of drug Only gives optimum values of , etc, not structure of new drug. Compounds must be described similarly even if structures are very different (using log P instead of can get round the problem - but then log P has to be measured (but does not as it is found in tables) Can prevent researchers looking at a new series of compounds Often have to use response from a single concentration rather than concentration to achieve set effect - therefore loss of accuracy A single QSAR only addresses one property - may need to consider solubility, stability, absorption, metabolism, transport, safety… QSAR only valid if all compounds in series operate by a common mechanism. This is often not valid. Requires accurate data on weakly active compounds.")
25
Molecular modelling Docking Geometry optimization/ Global minimum Energy distance Energy Conformation De novo ligand design /automatic docking

26
Rational Drug Discovery Molecular modelling Theory Drug structure – why is this important Drug conformation – how can you use knowledge of conformation in drug design Docking – different ways to do docking; ways to use it in drug design, limitations De Novo ligand design – role in drug design PC sessions – ideas is to illustrate the above

Similar presentations

 Workshop.>")
EVER WANTED TO KNOW Julia M. Goodfellow Dynamic Processes: Lecture 1 Lecture Notes.>")
: Reading: Van Holde, Chapter 1 Van Holde Chapter 3.1 to 3.3 Van Holde Chapter 2 (we’ll go through Chapters 1 and 3 first. 1.Van.>")









 Comparative Molecular Field Analysis (CoMFA) Gijs Schaftenaar.>")


 and Comparative Molecular Field Analysis (CoMFA) Martin Ott.>")
 Chen Yu Zong.>")

