Download presentation
Presentation is loading. Please wait.

1
Protein Structure Prediction Dr. G.P.S. Raghava Protein Sequence + Structure

2
Protein Structure Prediction Experimental Techniques –X-ray Crystallography –NMR Limitations of Current Experimental Techniques –Protein DataBank (PDB) -> 23000 protein structures –SwissProt -> 100,000 proteins –Non-Redudant (NR) -> 10,00,000 proteins Importance of Structure Prediction –Fill gap between known sequence and structures –Protein Engg. To alter function of a protein –Rational Drug Design

3
Different Levels of Protein Structure

5
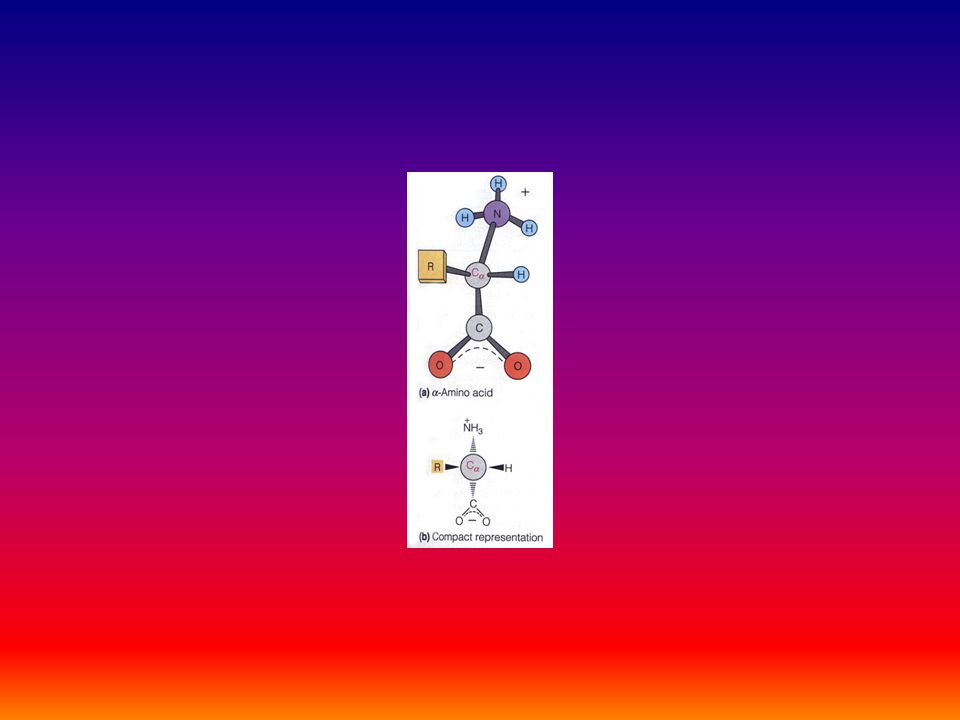
Protein Architecture Proteins consist of amino acids linked by peptide bonds Each amino acid consists of: –a central carbon atom –an amino group –a carboxyl group and –a side chain Differences in side chains distinguish the various amino acids

6
Amino Acid Side Chains Vary in: Size Shape Polarity

7
Peptide Bond

8
Peptide Bonds

9
Dihedral Angles

10
Conformation Flexibility Backbone (main chain of atoms in peptide bonds, minus side chains) conformation: –Torsion or rotation angles around: C-N bond ( ) C-C bond ( ) –Sterical hinderance: Most – Pro Least - Gly
 conformation: –Torsion or rotation angles around: C-N bond ( ) C-C bond ( ) –Sterical hinderance: Most – Pro Least - Gly")
11
Ramachandran Plot

12
Protein Secondary Structure Secondary Structure Regular Secondary Structure ( -helices, - sheets) Irregular Secondary Structure (Tight turns, Random coils, bulges)
 Irregular Secondary Structure (Tight turns, Random coils, bulges)")
13
Secondary Structure: Helices H-bond Individual Amino acid ALPHA HELIX : a result of H-bonding between every fourth peptide bond (via amino and carbonyl groups) along the length of the polypeptide chain
 along the length of the polypeptide chain")
15
Helix formation is local THYROID hormone receptor (2nll)
")
16
Secondary Structure: Beta Sheets BETA PLEATED SHEET: a result of H-bonding between polypeptide chains

17
-sheet formation is NOT local

18
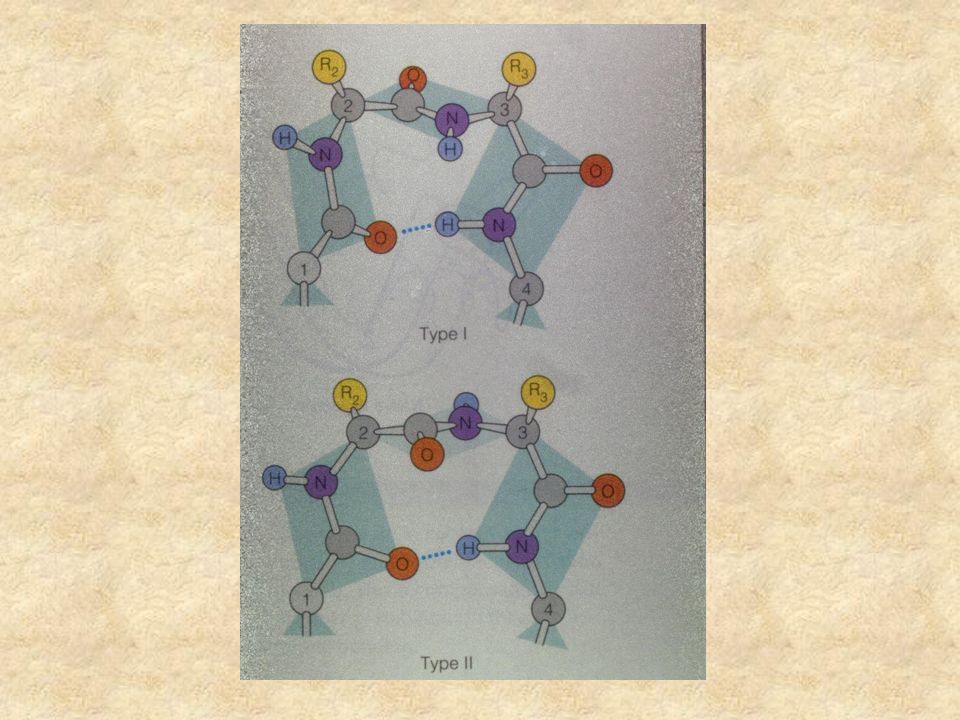
Definition of -turn A -turn is defined by four consecutive residues i, i+1, i+2 and i+3 that do not form a helix and have a C (i)-C (i+3) distance less than 7Å and the turn lead to reversal in the protein chain. (Richardson, 1981). The conformation of -turn is defined in terms of and of two central residues, i+1 and i+2 and can be classified into different types on the basis of and . i i+1 i+2 i+3H-bond D <7Å
. The conformation of -turn is defined in terms of and of two central residues, i+1 and i+2 and can be classified into different types on the basis of and . i i+1 i+2 i+3H-bond D <7Å.")
20
Tight turns TypeNo. of residuesH-bonding -turn 2 NH(i)-CO(i+1) -turn 3 CO(i)-NH(i+2) -turn 4 CO(i)-NH(i+3) -turn 5 CO(i)-NH(i+4) -turn 6 CO(i)-NH(i+5)
-CO(i+1) -turn 3 CO(i)-NH(i+2) -turn 4 CO(i)-NH(i+3) -turn 5 CO(i)-NH(i+4) -turn 6 CO(i)-NH(i+5).")
21
Secondary Structure shortcuts

22
Tertiary Structure: Hexokinase (6000 atoms, 48 kD, 457 amino acids) polypeptides with a tertiary level of structure are usually referred to as globular proteins, since their shape is irregular and globular in form
 polypeptides with a tertiary level of structure are usually referred to as globular proteins, since their shape is irregular and globular in form")
23
Quarternary Structure: Haemoglobin

24
What determines fold? Anfinsen’s experiments in 1957 demonstrated that proteins can fold spontaneously into their native conformations under physiological conditions. This implies that primary structure does indeed determine folding or 3-D stucture. Some exceptions exist –Chaperone proteins assist folding –Abnormally folded Prion proteins can catalyze misfolding of normal prion proteins that then aggregate

25
Levels of Description of Structural Complexity Primary Structure (AA sequence) Secondary Structure –Spatial arrangement of a polypeptide’s backbone atoms without regard to side-chain conformations , , coil, turns (Venkatachalam, 1968) –Super-Secondary Structure , , / , + (Rao and Rassman, 1973) Tertiary Structure –3-D structure of an entire polypeptide Quarternary Structure –Spatial arrangement of subunits (2 or more polypeptide chains)
 Secondary Structure –Spatial arrangement of a polypeptide’s backbone atoms without regard to side-chain conformations , , coil, turns (Venkatachalam, 1968) –Super-Secondary Structure , , / , + (Rao and Rassman, 1973) Tertiary Structure –3-D structure of an entire polypeptide Quarternary Structure –Spatial arrangement of subunits (2 or more polypeptide chains)")
26
Techniques of Structure Prediction Computer simulation based on energy calculation –Based on physio-chemical principles –Thermodynamic equilibrium with a minimum free energy –Global minimum free energy of protein surface Knowledge Based approaches –Homology Based Approach –Threading Protein Sequence –Hierarchical Methods

27
Energy Minimization Techniques Energy Minimization based methods in their pure form, make no priori assumptions and attempt to locate global minma. Static Minimization Methods –Classical many potential-potential can be construted –Assume that atoms in protein is in static form –Problems(large number of variables & minima and validity of potentials) Dynamical Minimization Methods –Motions of atoms also considered –Monte Carlo simulation (stochastics in nature, time is not cosider) –Molecular Dynamics (time, quantum mechanical, classical equ.) Limitations –large number of degree of freedom,CPU power not adequate –Interaction potential is not good enough to model
 Dynamical Minimization Methods –Motions of atoms also considered –Monte Carlo simulation (stochastics in nature, time is not cosider) –Molecular Dynamics (time, quantum mechanical, classical equ.) Limitations –large number of degree of freedom,CPU power not adequate –Interaction potential is not good enough to model.")
28
Molecular Dynamics Provides a way to observe the motion of large molecules such as proteins at the atomic level – dynamic simulation Newton’s second law applied to molecules Potential energy function –Molecular coordinates –Force on all atoms can be calculated, given this function –Trajectory of motion of molecule can be determined

29
Knowledge Based Approaches Homology Modelling –Need homologues of known protein structure –Backbone modelling –Side chain modelling –Fail in absence of homology Threading Based Methods –New way of fold recognition –Sequence is tried to fit in known structures –Motif recognition –Loop & Side chain modelling –Fail in absence of known example

30
Homology Modeling Simplest, reliable approach Basis: proteins with similar sequences tend to fold into similar structures Has been observed that even proteins with 25% sequence identity fold into similar structures Does not work for remote homologs (< 25% pairwise identity)
")
31
Homology Modeling Given: –A query sequence Q –A database of known protein structures Find protein P such that P has high sequence similarity to Q Return P’s structure as an approximation to Q’s structure

32
Threading Given: – sequence of protein P with unknown structure –Database of known folds Find: –Most plausible fold for P –Evaluate quality of such arrangement Places the residues of unknown P along the backbone of a known structure and determines stability of side chains in that arrangement

33
Hierarcial Methods Intermidiate structures are predicted, instead of predicting tertiary structure of protein from amino acids sequence Prediction of backbone structure –Secondary structure (helix, sheet,coil) –Beta Turn Prediction –Super-secondary structure Tertiary structure prediction Limitation Accuracy is only 75-80 % Only three state prediction
 –Beta Turn Prediction –Super-secondary structure Tertiary structure prediction Limitation Accuracy is only % Only three state prediction")
Similar presentations











![. Protein Structure Prediction [Based on Structural Bioinformatics, section VII]](/16/5116302/big_thumb.jpg ". Protein Structure Prediction [Based on Structural Bioinformatics, section VII]>")
 Today: Why bother about proteins/prediction Concepts of molecular.>")








