Download presentation
Presentation is loading. Please wait.

6
Aswad H. Al.Obeidy FICMS, FICMS GE&Hep Kirkuk General Hospital

7
Is a clonal disorder of a multipotent hematopoietic progenitor cell of unknown etiology characterized by Marrow fibrosis Extramedullary hematopoiesis Splenomegaly Chronic IMF is the least common chronic myeloproliferative disorder Establishing this diagnosis in the absence of a specific clonal marker is difficult because myelofibrosis and splenomegaly are also features of both PV and CML

8
Malignant Acute leukemia (lymphocytic, myelogenous, megakaryocytic) Chronic myelogenous leukemia Hairy cell leukemia Hodgkin disease Idiopathic myelofibrosis Lymphoma Multiple myeloma Myelodysplasia Polycythemia vera Systemic mastocytosis
 Chronic myelogenous leukemia Hairy cell leukemia Hodgkin disease Idiopathic myelofibrosis Lymphoma Multiple myeloma Myelodysplasia Polycythemia vera Systemic mastocytosis")
9
Nonmalignant HIV infection Hyperparathyroidism Renal osteodystrophy Systemic lupus erythematosus Tuberculosis Vitamin D deficiency Thorium dioxide exposure Gray platelet syndrome

10
The etiology of chronic IMF is unknown Nonrandom chromosome abnormalities such as 9p, 20q–, 13q–, trisomy 8 or 9, or partial trisomy 1q are common No cytogenetic abnormality specific to the disease has been identified The degree of myelofibrosis and the extent of extramedullary hematopoiesis are also not related Fibrosis in this disorder is associated with overproduction of transforming growth factor and tissue inhibitors of metalloproteinases Marrow angiogenesis occurs due to increased production of vascular endothelial growth factor (VEGF) Importantly, fibroblasts in chronic IMF are polyclonal and not part of the neoplastic clone
 Importantly, fibroblasts in chronic IMF are polyclonal and not part of the neoplastic clone")
11
No signs or symptoms are specific for chronic IMF Many patients are asymptomatic at presentation The disease is usually detected by the discovery of splenic enlargement and/or abnormal blood counts during a routine examination However, in contrast to its companion myeloproliferative disorders, night sweats, fatigue, and weight loss may be presenting complaints A blood smear shows the characteristic features of extramedullary hematopoiesis: teardrop-shaped red cells, nucleated red cells, myelocytes, and promyelocytes; myeloblasts may also be present Anemia, usually mild initially, is the rule, while the leukocyte and platelet counts are either normal or increased, but either can be depressed

12
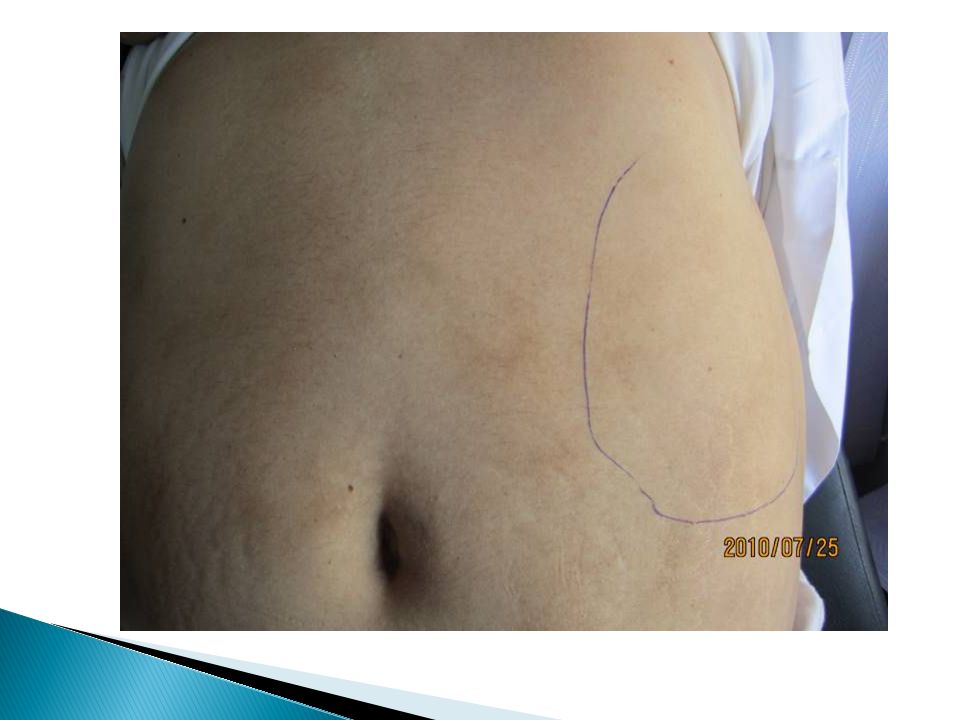
Mild hepatomegaly may accompany the splenomegaly but is unusual in the absence of splenic enlargement Isolated lymphadenopathy should suggest another diagnosis Both serum lactate dehydrogenase and alkaline phosphatase levels can be elevated The LAP score can be low, normal, or high. Marrow is usually inaspirable due to the myelofibrosis Bone x-rays may reveal osteosclerosis Exuberant extramedullary hematopoiesis can cause ascites, portal, pulmonary or intracranial hypertension, intestinal or ureteral obstruction, pericardial tamponade, spinal cord compression, or skin nodules Splenic enlargement can be sufficiently rapid to cause splenic infarction with fever and pleuritic chest pain Hyperuricemia and secondary gout may ensue

13
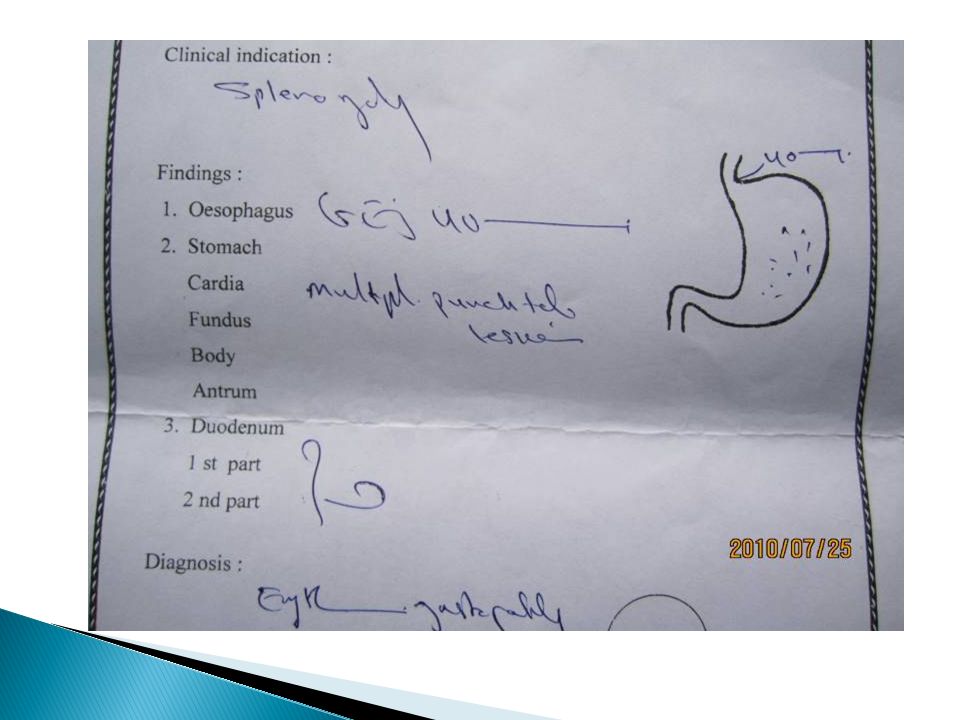
The diagnosis of primary myelofibrosis is actively considered when anemia or splenomegaly is accompanied by a “myelophthisic” blood picture consisting of immature granulocytes, nucleated red cells, and teardrop-shaped red blood cells (dacryocytes) on the peripheral blood smear Splenomegaly due to extramedullary hematopoiesis may be sufficiently massive to cause portal hypertension and variceal formation Cytogenetic analysis of blood is useful both to exclude CML and for prognostic purposes Approximately 45% of chronic IMF patients express the JAK2 V617F mutation had a poorer survival However, myelophthisis may also be associated with other bone marrow–infiltrating processes, including metastatic cancer, Hodgkin's disease and multiple myeloma Therefore, bone marrow biopsy is essential for further clarification of the diagnosis.
 on the peripheral blood smear Splenomegaly due to extramedullary hematopoiesis may be sufficiently massive to cause portal hypertension and variceal formation Cytogenetic analysis of blood is useful both to exclude CML and for prognostic purposes Approximately 45% of chronic IMF patients express the JAK2 V617F mutation had a poorer survival However, myelophthisis may also be associated with other bone marrow–infiltrating processes, including metastatic cancer, Hodgkin s disease and multiple myeloma Therefore, bone marrow biopsy is essential for further clarification of the diagnosis.")
14
The bone marrow in patients with primary myelofibrosis is not easily aspirated (dry tap) There are no characteristic morphologic abnormalities that distinguish IMF from the other chronic myeloproliferative disorders The usual findings in the core biopsy include atypical megakaryocyte hyperplasia, collagen fibrosis, osteosclerosis, and intrasinusoidal hematopoiesis The degree of fibrosis may be better estimated by the use of either a reticulin (silver impregnation) or trichrome stain Occasionally, the degree of bone marrow fibrosis may be minimal (“cellular phase” myeloid metaplasia) Bone marrow fibrosis may also be associated with other hematologic and nonhematologic conditions
 There are no characteristic morphologic abnormalities that distinguish IMF from the other chronic myeloproliferative disorders The usual findings in the core biopsy include atypical megakaryocyte hyperplasia, collagen fibrosis, osteosclerosis, and intrasinusoidal hematopoiesis The degree of fibrosis may be better estimated by the use of either a reticulin (silver impregnation) or trichrome stain Occasionally, the degree of bone marrow fibrosis may be minimal ( cellular phase myeloid metaplasia) Bone marrow fibrosis may also be associated with other hematologic and nonhematologic conditions")
15
Survival in chronic IMF varies according to specific clinical features but is shorter than in patients with PV or ET The natural history of chronic IMF is one of increasing marrow failure with transfusion-dependent anemia and increasing organomegaly due to extramedullary hematopoiesis As with CML, chronic IMF can evolve from a chronic phase to an accelerated phase with constitutional symptoms and increasing marrow failure About 10% of patients develop an aggressive form of acute leukemia for which therapy is usually ineffective Important prognostic factors for disease acceleration include anemia, leukocytosis, thrombocytopenia, the presence of circulating myeloblasts, older age, the presence of complex cytogenetic abnormalities, and constitutional symptoms such as unexplained fever, night sweats, or weight loss

16
No specific therapy exists for chronic IMF. Anemia may be due to gastrointestinal blood loss and exacerbated by folic acid deficiency, and in rare instances, pyridoxine therapy has been effective However, anemia is more often due to ineffective erythropoiesis uncompensated by extramedullary hematopoiesis in the spleen and liver Neither recombinant erythropoietin nor androgens, such as Danazol, have proved consistently effective as therapy for anemia Erythropoietin may worsen splenomegaly and will be ineffective if the serum erythropoietin level is >125 mU/L. A red cell splenic sequestration study can establish the presence of hypersplenism, for which splenectomy is indicated Splenectomy may also be necessary if splenomegaly impairs alimentation

17
Splenectomy should be performed before cachexia sets in. In this situation, splenectomy should not be avoided because of concern over rebound thrombocytosis, loss of hematopoietic capacity, or compensatory hepatomegaly. However, for unexplained reasons, splenectomy increases the risk of blastic transformation Splenic irradiation is, at best, temporarily palliative and associated with a significant risk of neutropenia and infection Allopurinol can control significant hyperuricemia Hydroxyurea has proved useful for controlling organomegaly in some patients The role of IFN- is still undefined and its side effects are more pronounced in the older individuals who are usually afflicted with this disorder Glucocorticoids have been used to control constitutional symptoms and autoimmune complications and may ameliorate anemia alone or in combination with low dose thalidomide (50–100 mg/d) Allogeneic bone marrow transplantation is the only curative treatment and should be considered in younger patients; reduced- intensity conditioning regimens may permit hematopoietic cell transplantation to be extended to older individuals
 Allogeneic bone marrow transplantation is the only curative treatment and should be considered in younger patients; reduced- intensity conditioning regimens may permit hematopoietic cell transplantation to be extended to older individuals.")
18
After splenectomy, acute leukemia, marked hepatomegaly, and extreme thrombocytosis may develop in approximately 16, 16, and 22% of surgical survivors, respectively Therefore, prophylactic hydroxyurea is strongly advised to prevent the post-splenectomy elevation in platelet count In poor surgical candidates with symptomatic splenomegaly, palliative splenic irradiation is reasonable (approximately 300 cGy given in 10 fractions) Splenic irradiation may also be used in the management of splenic infarcts that are refractory to opiate analgesics Treatment of nonsplenic extramedullary hematopoiesis requires low doses of involved field irradiation (100 to 150 cGy) given in multiple fractions
 Splenic irradiation may also be used in the management of splenic infarcts that are refractory to opiate analgesics Treatment of nonsplenic extramedullary hematopoiesis requires low doses of involved field irradiation (100 to 150 cGy) given in multiple fractions")
19
Allogeneic hematopoietic stem cell transplantation has provided durable disease remission in approximately a third of patients with primary myelofibrosis who have undergone such treatment However, the substantial mortality and morbidity (approximately 50%) associated with the procedure currently limit it to poor-risk young patients Lenalidomide, an immunomodulatory analogue of thalidomide, has therapeutic activity in primary myelofibrosis, and additional studies are ongoing In primary myelofibrosis, the immediate goal is to identify the causal genetic mutation
 associated with the procedure currently limit it to poor-risk young patients Lenalidomide, an immunomodulatory analogue of thalidomide, has therapeutic activity in primary myelofibrosis, and additional studies are ongoing In primary myelofibrosis, the immediate goal is to identify the causal genetic mutation")
20
The most frequent disease-related causes of death in patients with primary myelofibrosis are infection and leukemic transformation. The latter occurs in approximately 20% of patients over the first 10 years The presence of anemia (hemoglobin level less than 10 g/dL), thrombocytopenia (platelet count less than 100,000/μL), extreme ranges of white blood cell count (leukocytes either below 4000 or above 30,000/μL), circulating myeloblasts, unfavorable cytogenetic abnormalities (i.e., clones other than 13q– and 20q–), severe constitutional symptoms, or advanced age is predictive of poor survival A simple complete blood count–based prognostic scoring system reliably discriminates low-risk (median survival >10 years), intermediate-risk (median survival of 5 to 10 years), and high-risk disease (median survival <5 years) On the other hand, the degree of either hepatosplenomegaly or bone marrow fibrosis may not affect survival
, thrombocytopenia (platelet count less than 100,000/μL), extreme ranges of white blood cell count (leukocytes either below 4000 or above 30,000/μL), circulating myeloblasts, unfavorable cytogenetic abnormalities (i.e., clones other than 13q– and 20q–), severe constitutional symptoms, or advanced age is predictive of poor survival A simple complete blood count–based prognostic scoring system reliably discriminates low-risk (median survival >10 years), intermediate-risk (median survival of 5 to 10 years), and high-risk disease (median survival <5 years) On the other hand, the degree of either hepatosplenomegaly or bone marrow fibrosis may not affect survival.")
Similar presentations







II Dr. Ibrahim. A. Adam.>")


 Myelodysplastic / myeloproliferative diseases (MDS/MPD) >")

>")
 DEFINITION CLL is a neoplastic disease characterized by proliferation and accumulation (blood, marrow and lymphoid.>")

:>")
 leukemia Is characterized by an unregulated proliferation of myeloid elements in the bone marrow,>")




 - Essential Thrombocythemia - Myelofibrose Myeloid Methaplasia.>")
