Download presentation
Presentation is loading. Please wait.

1
Sequence Alignment and Phylogenetic Prediction using Map Reduce Programming Model in Hadoop DFS Presented by C. Geetha Jini (07MW03) D. Komagal Meenakshi (07MW05) Guided by Dr. G. Sudha Sadasivam Asst. Professor Dept. of CSE
 D. Komagal Meenakshi (07MW05) Guided by Dr. G. Sudha Sadasivam Asst. Professor Dept. of CSE.")
2
What is Sequence Alignment? The procedure of comparing two or more sequences by searching for a series of individual characters or character patterns that are in the same order in the sequences.

3
Types of Sequence Alignment Pair-wise Alignment Alignment of two sequences Global –using Needleman Wunsch algorithm. L G P S S K Q T G K G S _ S R A W D N | | | | | | | L N _ A T K S A G K G A I M R L G D A Local – using Smith Waterman algorithm. _ _ _ _ _ _ _ _ _ T G K G _ _ _ _ _ _ _ _ _ _ | | | _ _ _ _ _ _ _ _ _ A G K G _ _ _ _ _ _ _ _ _ _ Multiple Sequence Alignment Alignment of more than two sequences

4
Initialization F(0, 0) = 0 F(0, i) = −i * d F(j, 0) = −j* d Main Iteration For each i=1…M and j=1….N F(i-1,j-1+s(x i,y j ), case 1 F(i,j) = max F(i-1,j)-d, case 2 F(I,j-1)-d, case 3 DIAG, if case 1 Ptr(i,j) = UP, if case 2 LEFT, if case 3 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap NEEDLEMAN WUNSCH ALGORITHM s(x i,y j ) = +1, match -1, mismatch
 = 0 F(0, i) = −i * d F(j, 0) = −j* d Main Iteration For each i=1…M and j=1….N F(i-1,j-1+s(x i,y j ), case 1 F(i,j) = max F(i-1,j)-d, case 2 F(I,j-1)-d, case 3 DIAG, if case 1 Ptr(i,j) = UP, if case 2 LEFT, if case 3 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap NEEDLEMAN WUNSCH ALGORITHM s(x i,y j ) = +1, match -1, mismatch")
5
Needleman Wunsch Algorithm AGTA 0 -2-3-4 A1 0 -2 T 00 1 0 A-3 02 F(i,j) i=0 1 2 3 4 j=0 1 2 3 f(0,0)+s(1,1) =1 F(1,1)=max f(0,1)-1 = -2 f(1,0)-1 = -2 = 1 (case 1) OptimalAlignment A_TA AGTA Score:1+0+1+2 = 4 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap s(x i,y j ) = +1, match -1, mismatch d=1 PTR = DIAG, if case 1 UP, if case 2 LEFT, if case 3 f(0,1)+s(1,2) =-2 f(0,2)-1 = -3 f(1,1)-1 = 0 Max = 0 (case 3)
 i= j= f(0,0)+s(1,1) =1 F(1,1)=max f(0,1)-1 = -2 f(1,0)-1 = -2 = 1 (case 1) OptimalAlignment A_TA AGTA Score: = 4 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap s(x i,y j ) = +1, match -1, mismatch d=1 PTR = DIAG, if case 1 UP, if case 2 LEFT, if case 3 f(0,1)+s(1,2) =-2 f(0,2)-1 = -3 f(1,1)-1 = 0 Max = 0 (case 3)")
6
Smith Waterman Algorithm Initialization: F(0, j) = F(i, 0) = 0 Iteration: 0 F(i, j) = max F(i – 1, j – 1) + s(x i, y j ), case 1 F(i – 1, j) – d, case 2 F(i, j – 1) – d, case 3
 = F(i, 0) = 0 Iteration: 0 F(i, j) = max F(i – 1, j – 1) + s(x i, y j ), case 1 F(i – 1, j) – d, case 2 F(i, j – 1) – d, case 3")
7
Smith Waterman Algorithm AGTA 0 0000 A 0 1 00 0 T000 1 0 A 0 0002 F(i,j) i=0 1 2 3 4 j=0 1 2 3 f(0,0)+s(1,1) =1 F(1,1)=max f(0,1)-1 = -1 f(1,0)-1 = -1 0 = 1 (case 1) OptimalAlignment A_TA _ _TA Score: 1+2 = 4 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap s(x i,y j ) = +1, match -1,mismatch d=1 PTR = DIAG, if case 1 UP, if case 2 LEFT, if case 3 f(0,2)+s(1,3) =-1 F(1,3)=max f(0,3)-1 = -1 f(1,2)-1 = -1 0 = 0
 i= j= f(0,0)+s(1,1) =1 F(1,1)=max f(0,1)-1 = -1 f(1,0)-1 = -1 0 = 1 (case 1) OptimalAlignment A_TA _ _TA Score: 1+2 = 4 Case 1: x i aligns to y i Case 2: x i aligns to gap Case 3: y i aligns to gap s(x i,y j ) = +1, match -1,mismatch d=1 PTR = DIAG, if case 1 UP, if case 2 LEFT, if case 3 f(0,2)+s(1,3) =-1 F(1,3)=max f(0,3)-1 = -1 f(1,2)-1 = -1 0 = 0")
8
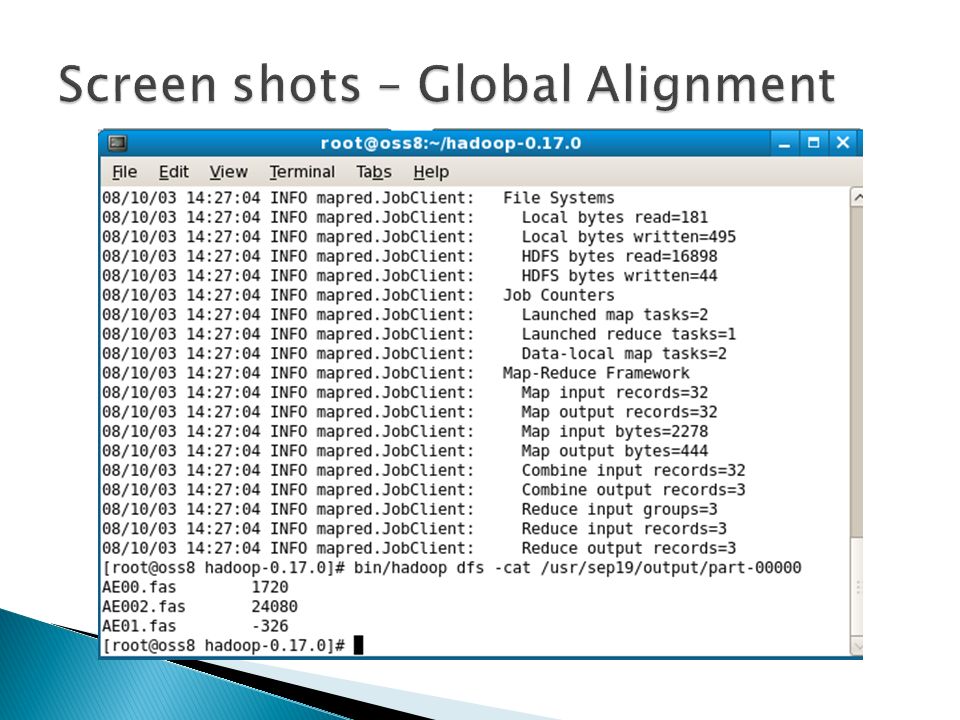
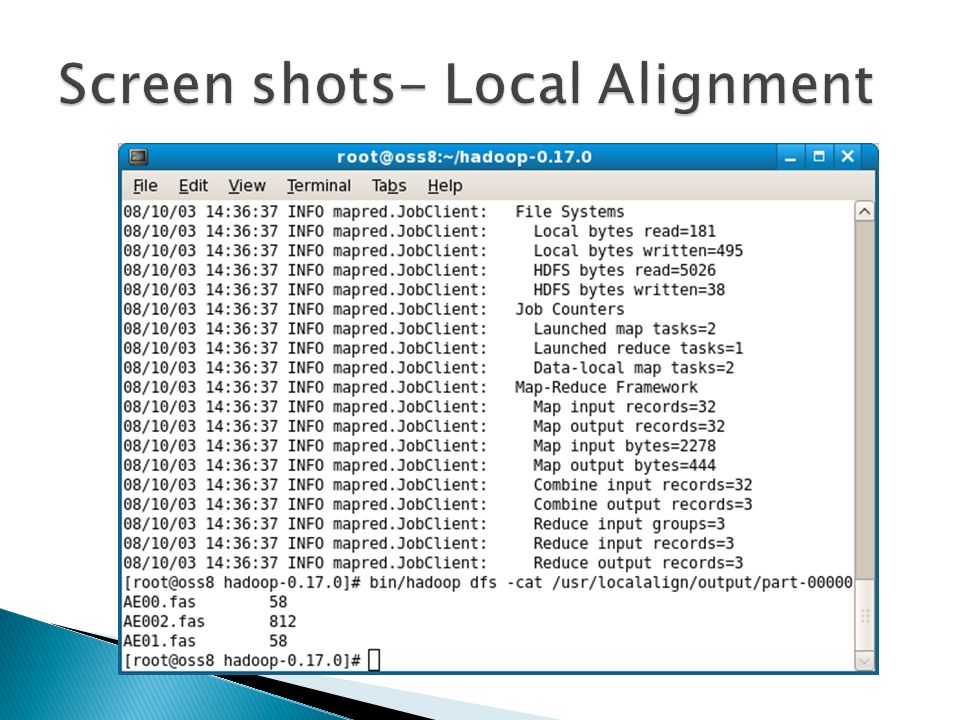
Input: one query file and a set of sequence files Put all files in DFS Map Reduce Combine all the (K,V) pairs Output: (Filename, Score) Set File Name as Key Pass Entire File contents as Value Do Sequence alignment of query file with the target files in DFS Return (Filename as key, Score as Value). Proposed system

9
A multiple sequence alignment is a sequence alignment of three or more biological sequences, generally protein, DNA, or RNA. In general, the input is a set of query sequences that are assumed to have an evolutionary relationship by which they share a lineage and are descended from a common ancestor. From the resulting multiple sequence alignment, phylogenetic analysis can be conducted to assess the sequences shared evolutionary origins.

10
Dynamic programming Progressive alignment construction Methods for producing MSA

11
most direct method for producing an MSA to identify the globally optimal alignment solution. computational complexity ◦ For n individual sequences, the naive method requires constructing the n-dimensional equivalent of the matrix formed in standard pairwise sequence alignment. ◦ The search space thus increases exponentially with increasing n and is also strongly dependent on sequence length.

12
uses a heuristic search. builds up a final MSA by combining pair wise alignments beginning with the most similar pair and progressing to the most distantly related. The most popular progressive alignment method has been the ClustalW. All progressive alignment methods require two stages: ◦ a first stage in which the relationships between the sequences are represented as a tree, called a guide tree. ◦ second step in which the MSA is built by adding the sequences sequentially to the growing MSA according to the guide tree.

13
◦ first step: computation of guide tree from pair-wise alignment scores by an efficient clustering method such as neighbor-joining method. ◦ Second step: The two most similar sequences are aligned first, additional sequences (or groups of sequences) are added later following the guide tree ◦ requires a method to optimally align a sequence with an alignment or an alignment with an alignment sequence 1 sequence 2 sequence 3 Sequence4 Example: According to guide tree, align first sequences 1 and 2, then align sequence 3 to alignment of sequence 1 and 2, then sequence 4 to alignment of sequences 1, 2, and 3.
 are added later following the guide tree ◦ requires a method to optimally align a sequence with an alignment or an alignment with an alignment sequence 1 sequence 2 sequence 3 Sequence4 Example: According to guide tree, align first sequences 1 and 2, then align sequence 3 to alignment of sequence 1 and 2, then sequence 4 to alignment of sequences 1, 2, and 3..")
14
Neighbor-joining is a bottom-up clustering method used for the construction of phylogenetic trees. Neighbor-joining is an iterative algorithm. Each iteration consists of the following steps: Based on the current distance matrix calculate the matrix Q. For example, if we have four taxa (A, B, C, D) and the following distance matrix:
 and the following distance matrix:.")
15
We obtain the following values for the Q matrix: Find the pair of taxa in Q with the lowest value. Create a node on the tree that joins these two taxa (i.e. join the closest neighbors, as the algorithm name implies).
..")
16
Calculate the distance of each of the taxa in the pair to this new node. Calculate the distance of all taxa outside of this pair to the new node. Start the algorithm again, considering the pair of joined neighbors as a single taxon and using the distances calculated in the previous step.

17
The primary problem is that when errors are made at any stage in growing the MSA, these errors are then propagated through to the final result. Performance is also particularly bad when all of the sequences in the set are rather distantly related.

18
Phylogenetic Analysis An investigation of evolutionary relationships among a group of related sequences by producing a tree representation of relationships. Significant use-to make prediction concerning tree of life.

19
Structure outer branches ->Sequences Inner part -> Reflect the degree to which sequences are related Alike sequences -> located at neighboring outside branches Less related sequences -> more distant from each other

20
Proposed System Implementation of Sequence alignment and phylogenetic prediction using map-reduce programming model in hadoop Algorithms used for Alignment Global-Needleman Wunsch Algorithm Local-Smith Waterman Algorithm

21
Input: set of sequence files Put all files in DFS Map Reduce Combine all the (K,V) pairs Output: (Filename, Score) Phylogenetic Analysis Set File Name as Key Pass Entire File contents as Value Do Sequence alignment of all the files with all possible combinations and find the alignment scores Return (Filename as key, Score as Value). Proposed system

24
The mapreduce algorithm for pairwise sequence alignment both local and global was completed using the Needleman wunsch and Smith waterman algorithm in Hadoop. This can be extended to do multiple sequence alignment and to perform phylogenetic analysis in Hadoop for predicting possible evolutionary relationships among a group of related sequences.

25
Bibliography David W. Mount, Bioinformatics Sequence and Genome Analysis, second edition http://apache.org/hadoop http://wiki.apache.org/hadoop Map reduce: Simplified data processing on Large Clusters, Jeffrey Dean and Sanjay Ghemawat www.biojava.org www.biojava.org/wiki/Biojava:CookBook Biojava in Anger, A Tutorial and Recipe for Those in a Hurry. www.di.unito.it/~botta/didattica/biojavaHowTo.pdf http://www-sop.inria.fr/oasis/Stages/04-05/BioProActive-Caromel.html http://hpc.pnl.gov/projects/scalablast/ http://www.ebi.ac.uk/Tools/clustalw2/

26
Thank you

Similar presentations
*>")






 Modified by Benny Chor, from slides by Shlomo Moran and Ydo Wexler (IIT)>")











