Download presentation
Presentation is loading. Please wait.

1
A highly abbreviated introduction to proteomics

2
A typical shotgun proteomics experiment
Collect tens of thousands of MS/MS spectra Can identify >1,000 proteins from cell lysate

3
Orbi video:

4
Shotgun proteomics identifies proteins from the fragmentation mass spectra of their constituent peptides b & y ions Peptide fragmentation Actual peptide tandem (MS/MS) mass spectrum Idealized peptide tandem (MS/MS) mass spectrum from database Idealized peptide tandem (MS/MS) mass spectrum with PTM (phosphoserine) Marcotte (2007) Nature Biotechnology 25:
 mass spectrum. Idealized peptide. tandem (MS/MS) mass spectrum. from database. Idealized peptide. tandem (MS/MS) mass spectrum. with PTM. (phosphoserine) Marcotte (2007) Nature Biotechnology 25:")
5
One common strategy for relative quantification =
using isotopically labeled samples (e.g. 15N vs. 14N, 13C vs. 12C, etc.) SILAC = stable isotope labeling with amino acids in cell culture iCAT = isotope tags on cysteines iTRAQ = isobaric labels on cysteines (same mass, different isotopes) AQUA = absolute quantification by spiking in isotopically shifted peptide standards for proteins of interest Mallick & Kuster (2010) Nature Biotechnology 28:
 SILAC = stable isotope labeling with. amino acids in cell culture. iCAT = isotope tags on cysteines. iTRAQ = isobaric labels on cysteines. (same mass, different isotopes) AQUA = absolute quantification by spiking. in isotopically shifted peptide. standards for proteins of interest. Mallick & Kuster (2010) Nature Biotechnology 28:")
6
Mass spectrometry strategies for measuring absolute protein abundances
for 100’s to 1000’s of proteins adapted from Vogel & Marcotte Nature Biotechnology , 825-6

7
& the current state-of-the-art …
Each K peptides, from ~10,000 proteins spanning ~7 orders of magnitude in abundance

8
A highly abbreviated introduction to
large-scale protein interaction screens

10
X-ray structure of ATP synthase Schematic version Network representation a b g d b2 e a c12 Total set = protein complex Sum of direct + indirect interactions

12
High-throughput yeast two-hybrid
+ DBD Bait Prey Act DNA binding domain Transcription activation domain Prey Act Core transcription machinery Bait DBD transcription operator or upstream activating sequence Reporter gene

13
High-throughput yeast two-hybrid
Haploid yeast cells expressing activation domain- prey fusion proteins Diploid yeast probed with DNA-binding domain- Pcf11 bait fusion protein

14
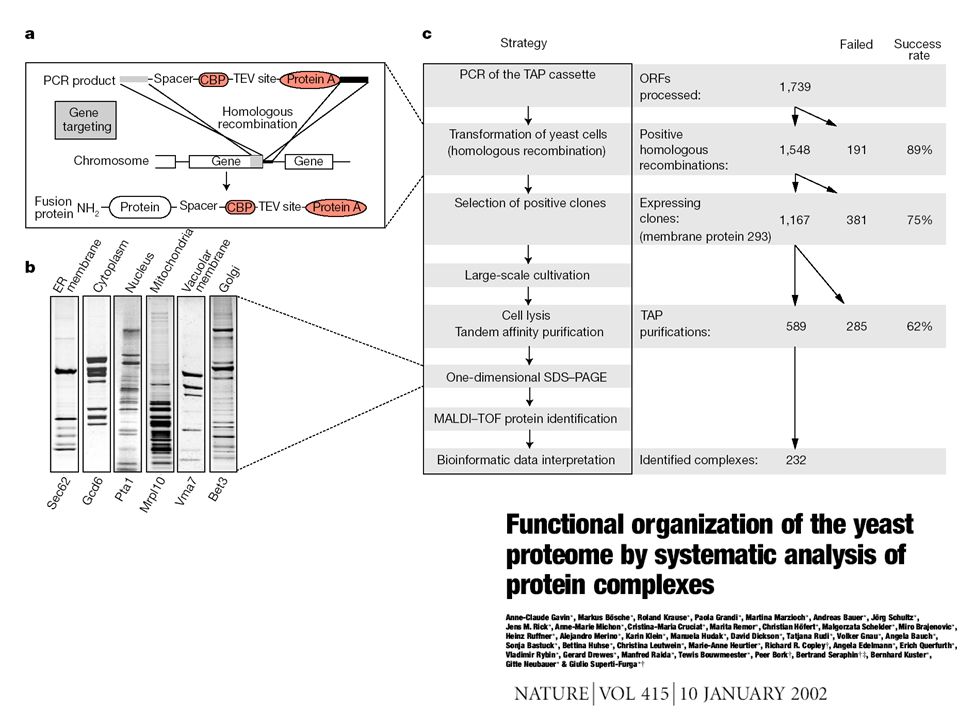
High-throughput complex mapping by mass spectrometry
Tag Bait Affinity column protein 1 protein 2 SDS- page protein 3 Trypsin digest, identify peptides by mass spectrometry protein 4 protein 5 protein 6

15
493 bait proteins 3617 “interactions”

16
A variant: tandem affinity purification (TAP)
Tag1 Tag2 Bait Affinity column2 protein 1 Affinity column1 protein 2 SDS- page protein 3 protein 4 + protease protein 5 protein 6 Trypsin digest, identify peptides by mass spectrometry Affinity column1

18
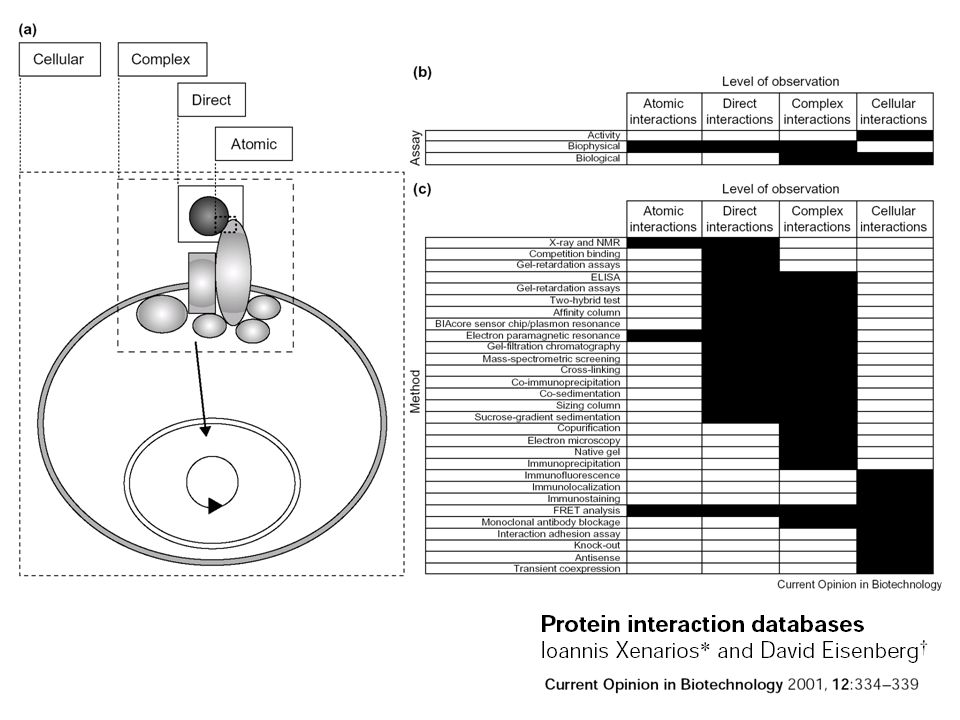
Estimating accuracy with a well-determined reference set of interactions

19
Where we were, more or less, until recently in terms of PPI maps

20
The current state-of-the-art in animal PPI maps
~3,500 affinity purification experiments ~11K interactions / ~2.3K proteins spans 556 complexes Still daunting for the human proteome Guruharsha et al. (2011) Cell 147, 690–703
 Cell 147, 690–703.")
21
>2,000 biochemical fractions,
Finding stable protein assemblies by native separations and quantitative mass spec. >2,000 biochemical fractions, including replicates >9,000 hours mass spec machine time Havugimana, Hart, et al., Cell (2012)
")
22
The profiles cover > ½ the experimentally verified proteome
& proteins within the same stable complexes co-elute Havugimana, Hart, et al., Cell (2012)
")
23
Turning separations into complexes
1) One separation, #13 of many Cluster 4) Inferred complexes ~5600 proteins ~120 fractions ... 59 60 61 62 63 64 Co-separation of the exocyst complex exoc1 exoc2 exoc3 exoc4 exoc5 exoc6 exoc7 exoc8 3) Inferred interactions high correlation >> more likely in complex 2) Pairwise protein correlations Machine learning (SVM, Ensemble methods) Hurdle is false positives: since you’re searching the entire space of possible shared complex memberships, where true co-memberships are extremely sparse, high scorers are dominated by false positives. Must use external data to 2b) External data Co-expression, shared protein domains, much more (HumanNet) Other AP-MS datasets (Guruharsha 2011, Malovannaya 2011)
 One separation, #13 of many. Cluster. 4) Inferred complexes. ~5600 proteins. ~120. fractions Co-separation of the exocyst complex. exoc1. exoc2. exoc3. exoc4. exoc5. exoc6. exoc7. exoc8. 3) Inferred interactions. high correlation >> more likely in complex. 2) Pairwise protein correlations. Machine learning. (SVM, Ensemble methods) Hurdle is false positives: since you’re searching the entire space of possible shared complex memberships, where true co-memberships are extremely sparse, high scorers are dominated by false positives. Must use external data to. 2b) External data. Co-expression, shared protein domains, much more (HumanNet) Other AP-MS datasets (Guruharsha 2011, Malovannaya 2011)")
24
Guiding and testing the reconstruction with known complexes
Havugimana, Hart, et al., Cell (2012)
")
25
13,998 high-confidence physical interactions / 3,011 proteins
A reference map of human protein complexes 13,998 high-confidence physical interactions / 3,011 proteins Defines >600 complexes: >100 heterodimers, >500 with ≥3 components Havugimana, Hart, et al., Cell (2012)
")
26
In yeast, phenotypes reflect biological modules.
e.g., lethality is tied not to the protein, but to the molecular machine small nucleolar ribonucleoprotein complex SAGA transcription factor/ chromatin remodeling complex TAFIID complex protein phosphatase 2A complex Essential gene Nonessential gene Hart, Lee, & Marcotte, BMC Bioinformatics 8:236 (2007)
")
27
The human protein complexes are also strongly enriched
for genes linked to the same diseases and phenotypes Havugimana, Hart, et al., Cell (2012)
")
28
The complexes are strongly enriched for genes linked to the same diseases, e.g., as for Cornelia de Lange Syndrome prweb.com Now confirmed by Deardorff et al., Am. J. Hum. Genet. 90, 1014–1027 Dermatology Online Journal 7(2): 8
: 8.")
29
Our current state of the art animal complex map
Cuihong Wan Blake Borgeson w/ Andrew Emili’s lab

30
Our current state of the art animal complex map
Extending the map Now 7 animals, >65 separations, nearly 7,000 mass spec experiments >3,500 fractions ~12,000 proteins Our current state of the art animal complex map >3,000 fractions ~9,000 proteins Cuihong Wan Blake Borgeson w/ Andrew Emili’s lab

Similar presentations


 and by size (SDS-PAGE) 2D-gel electrophoresis & mass spectrometry 3. Peptide.>")
 Data Kay Hofmann – Protein Evolution Group Week 5: Proteomics.>")






 Functional Genomics TranscriptomicsRNA Proteomics PROTEIN Metabolomics METABOLITE Transcription Translation Enzymatic.>")






 Course Director David Fenyö Contact information>")
 Course Director David Fenyö Contact information>")
 PNAS 100(12), 6940-6945. presented by Jessica.>")