Download presentation
Presentation is loading. Please wait.

1
NGS data analyses with BioUML Fedor Kolpakov Biosoft.Ru, Ltd. Institute of Systems Biology, Ltd. Novosibirsk, Russia

2
Agenda BioUML overview NGS tools – quality control – alignment tools – annotation tools – workflows Genome browser Archakov’s genome Ribosome profiling Live demonstration

3
BioUML overview

4
BioUML platform BioUML is an open source integrated platform for systems biology that spans the comprehensive range of capabilities including access to databases with experimental data, tools for formalized description, visual modeling and analyses of complex biological systems. Due to scripts (R, JavaScript) and workflow support it provides powerful possibilities for analyses of high- throughput data. Plug-in based architecture (Eclipse run time from IBM is used) allows to add new functionality using plug-ins. BioUML platform consists from 3 parts: BioUML server – provides access to biological databases; BioUML workbench – standalone application. BioUML web edition – web interface based on AJAX technology;
 and workflow support it provides powerful possibilities for analyses of high- throughput data. Plug-in based architecture (Eclipse run time from IBM is used) allows to add new functionality using plug-ins. BioUML platform consists from 3 parts: BioUML server – provides access to biological databases; BioUML workbench – standalone application. BioUML web edition – web interface based on AJAX technology;.")
5
Main platforms for bioinformatics and BioUML Taverna standalone application powerful workflows Galaxy workflows, web interface, collaborative research, genome browser scripts, statistics, plots R/Bioconductor BioUML platform standalone application powerful workflows web interface, collaborative research genome browser scripts, statistics, plots BioClipse Eclipse plug-in based architecture, chemoinformatics Eclipse plug-in based architecture, chemoinformatics

6
Main platforms for bioinformatics and BioUML Taverna standalone application powerful workflows Galaxy workflows, web interface, collaborative research, genome browser scripts, statistics, plots R/Bioconductor BioUML platform standalone application powerful workflows web interface, collaborative research genome browser scripts, statistics, plots + systems biology visual modelling simulation parameters fitting … + chat for on-line consultations BioClipse Eclipse plug-in based architecture, chemoinformatics Eclipse plug-in based architecture, chemoinformatics

7
Android market Android AppStore MacOS, iPOD, iPhone Market Platform Biostore BioUML

8
Biostore BioUML platform Developers - plug-ins: methods, visualization, etc. - databases Users - subscriptions - collaborative & reproducible research Experts -services for data analysis - on-line consultations BioUML ecosystem provide tools and databases use provide services

9
NGS - интегрированные в BioUML методы (Bowtie, MACS, ChIPHorde, ChIPMunk, …) - программы, интегрированные в Galaxy - пакеты R - аннотация найденных пиков (SNP, сайтов и т.п.) - визуализация - workflows - ChIP-SEQ - RNA-SEQ - сборка и аннотация генома человека (в процессе) - поддержка распарелеливания внешних программ как часть workflow - база данных GTRD (на основе данных ChIP-SEQ) - выделенные сервера - Amazon EC2 – по запросу - Biodatomics – 64 ядра, 256 Гб памяти.
 - программы, интегрированные в Galaxy - пакеты R - аннотация найденных пиков (SNP, сайтов и т.п.) - визуализация - workflows - ChIP-SEQ - RNA-SEQ - сборка и аннотация генома человека (в процессе) - поддержка распарелеливания внешних программ как часть workflow - база данных GTRD (на основе данных ChIP-SEQ) - выделенные сервера - Amazon EC2 – по запросу - Biodatomics – 64 ядра, 256 Гб памяти.")
12
Galaxy – analyses methods

13
Galaxy - workflow

14
Raw data preprocessing Track statistics Gather various statistics about track or FASTQ file Preprocess raw reads Remove reads not satisfying simple quality tests, removes adapters, trims low quality bases from read ends

15
Bowtie - fast - no indels - used for chip-seq Novoalign -single-end and paired-end - in nucleotide and color space - handle indels, - finds global optimum alignments using full Needleman-Wunsch algorithm выравнивание коротких ридов:

16
RNA-seq with tophat and Cuff* tools

17
ChIP-seq Bowtie Bowtie for alignment MACS MACS for peak calling ChipMunkIPSMEME ChipMunk, IPS, MEME for motif discovery

18
Popular NGS toolboxes available: GATK, Picard, SAM tools

19
An example: workflow for analyses of ChIP-Seq data

20
example: RNA-seq workflow

21
NGS data quality control 2 examples: rna-seq data (rat, IPS ) genome data – Archakov’s genome
 genome data – Archakov’s genome")
22
Track statistics (FastQC) Estimate quality of RAW or aligned reads like in FastQC program http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ All original FastQC processors are supported Works faster than FastQC Additional processor: Overrepresented prefixes Overrepresented K-mers works more precise (do not skip 80% of sequences) Along with HTML report separate statistics tables are generated and accessible for further analysis Ability to merge several reports into composite report As any BioUML analysis can become a part of workflow, scripts, etc. Tested on Archakov AP3 (RAW reads: 5.9Gb csfasta+12.7Gb qual), analysis time: 36 min (all processors) Tested on Zakian db50 (RAW reads: 6.5Gb fastq), analysis time: 7 min (all processors)
, analysis time: 36 min (all processors) Tested on Zakian db50 (RAW reads: 6.5Gb fastq), analysis time: 7 min (all processors).")
23
Track statistics launch Input data: BAM, FastQ and Solid (colorspace) data supported Whether reads should be aligned by left or right side Switch off individual processors to save time.
 data supported Whether reads should be aligned by left or right side Switch off individual processors to save time.")
24
Track statistics results (Archakov AP3): Quality per base
: Quality per base")
25
Track statistics results (Archakov AP3): Quality per sequence
: Quality per sequence")
26
Track statistics results (Archakov AP3): Nucleotide content per base
: Nucleotide content per base")
27
Track statistics results (Archakov AP3): GC content per base
: GC content per base")
28
Track statistics results (Archakov AP3): GC content per sequence
: GC content per sequence")
29
Track statistics results (Archakov AP3): N content per base
: N content per base")
30
Track statistics results (Archakov AP3): Duplicate sequences
: Duplicate sequences")
31
Track statistics results (Archakov AP3): Overrepresented sequences and 5-mers
: Overrepresented sequences and 5-mers")
32
Track statistics results (Archakov AP3): Overrepresented prefixes
: Overrepresented prefixes")
33
Track statistics results (Zakian db50): Quality per base
: Quality per base")
34
Track statistics results (Zakian db50): Quality per sequence
: Quality per sequence")
35
Track statistics results (Zakian db50): Nucleotide content per base
: Nucleotide content per base")
36
Track statistics results (Zakian db50): GC content per base
: GC content per base")
37
Track statistics results (Zakian db50): GC content per sequence
: GC content per sequence")
38
Track statistics results (Zakian db50): N content per base
: N content per base")
39
Track statistics results (Zakian db50): Duplicate sequences
: Duplicate sequences")
40
Track statistics results (Zakian db50): Overrepresented sequences and 5-mers
: Overrepresented sequences and 5-mers")
41
Genome browser

42
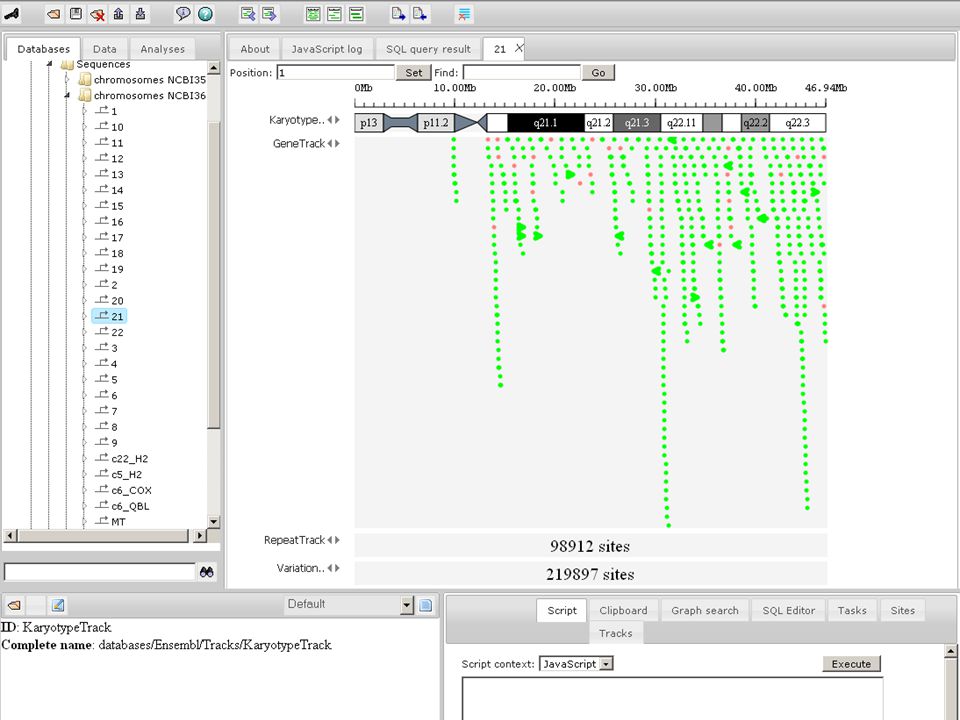
uses AJAX and HTML5 technologies interactive - dragging, semantic zoom tracks support Ensembl DAS-servers user-loaded BED/GFF/Wiggle files Genome browser: main features

43
DAS The Distributed Annotation System (DAS) defines a communication protocol used to exchange annotations on genomic or protein sequences.annotations It is motivated by the idea that such annotations should not be provided by single centralized databases, but should instead be spread over multiple sites. Data distribution, performed by DAS servers, is separated from visualization, which is done by DAS clients. DAS is a client-server system in which a single client integrates information from multiple servers. It allows a single machine to gather up sequence annotation information from multiple distant web sites, collate the information, and display it to the user in a single view. DAS is heavily used in the genome bioinformatics community. Over the last years we have also seen growing acceptance in the protein sequence and structure communities.

46
Genome browser Two BAM tracks are compared with each other (Example view on Human NCBI37 Chr.1) Profile is visible showing the coverage
 Profile is visible showing the coverage")
47
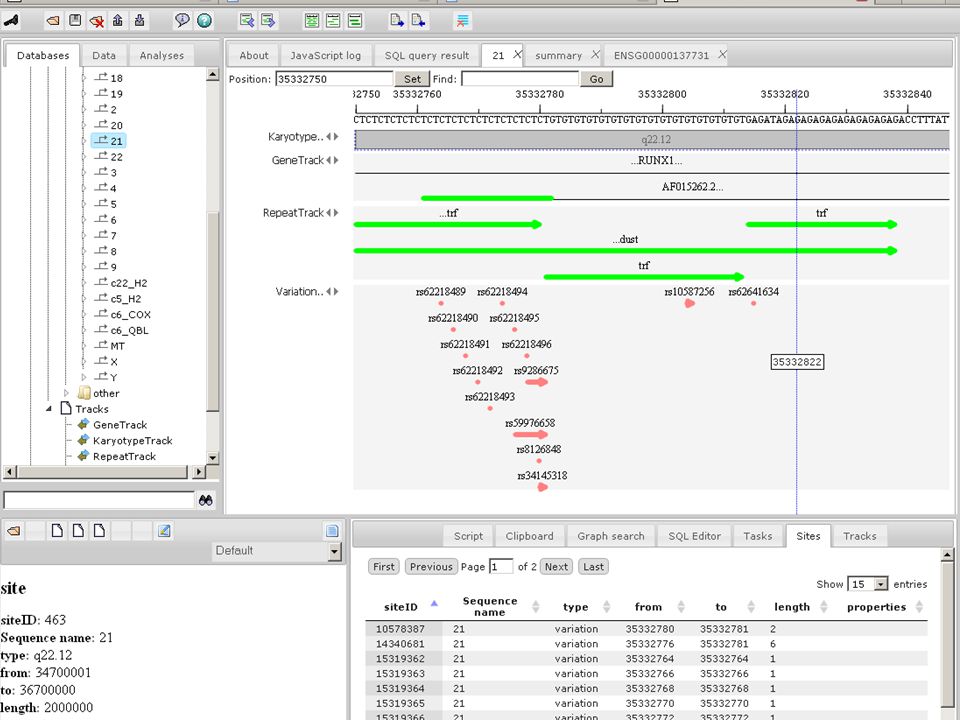
Genome browser Upon zooming individual reads become visible. All information associated with selected read is displayed in the Info box

48
Genome browser In detailed scale phred qualities graph is displayed along with changed nucleotides between read and reference sequence

50
NGS data Archakov’s genome

51
Preprocessing 1.Remove duplicates Purpose is to mitigate the effects of PCR amplification bias introduced during library construction. Two read pairs considered duplicate if they align to the same genomic position. >60% were removed as duplicates Alignments after this step: 213 531 460

52
Preprocessing 2. Local realignment Read mapping algorithms operate on each read independently, locally realign reads such that the number of mismatching bases is minimized across all the reads.

53
Preprocessing 3. Remove duplicates after realignment Realignment may change genomic positions of read pairs, after this step additional duplicates can be identified. 712 reads were removed (<0.00035%)
.")
54
Preprocessing 4. Recalibration of base quality values For each base in each read calculates various covariates (such as reported quality score, cycle, dinucleotide, GC-content). Using these values build the model that predicts sequencing errors. Then apply this model to calculate an empirical base quality score and overwrites the phred quality score currently in the read.
. Using these values build the model that predicts sequencing errors. Then apply this model to calculate an empirical base quality score and overwrites the phred quality score currently in the read..")
55
Genotyping 1.Call SNV by GATK 'Unified Genotyper' 2.Assign a well-calibrated probability to each variant call. Estimate the probability that SNV is a true genetic variant versus a sequencing or data processing artifact given SNP call annotations provided by 'Unuified Genotyper' (DepthOfCoverage, StrandBias, HaplotypeScore, ReadPosRankSumTest for example). o Variant Annotator - create the set of "true variants" from dbSNP, Hapmap and 1000 genomes databases. o Variant Recalibrator - create a Gaussian mixture model by looking at the annotations values over a high quality subset of the input call set ("true variants"). o Apply Variant Recalibration - apply the model parameters to each variant identified by Unified Genotyper calculating log odds ratio of being a true variant versus being false under the trained Gaussian mixture model.
. o Variant Annotator - create the set of true variants from dbSNP, Hapmap and 1000 genomes databases. o Variant Recalibrator - create a Gaussian mixture model by looking at the annotations values over a high quality subset of the input call set ( true variants ). o Apply Variant Recalibration - apply the model parameters to each variant identified by Unified Genotyper calculating log odds ratio of being a true variant versus being false under the trained Gaussian mixture model..")
56
Genotyping 3. Call indels by GATK 'Unified Genotyper' 4. Assign a well-calibrated probability to each indel. Similar to SNV calling but use only indels from 1000 Genomes as "true variants"

57
Genotyping 5. Filter out low quality variant calls. 1 783 656 SNVs 17 110 Indels 6. Annotate identified variants relative to genes.

58
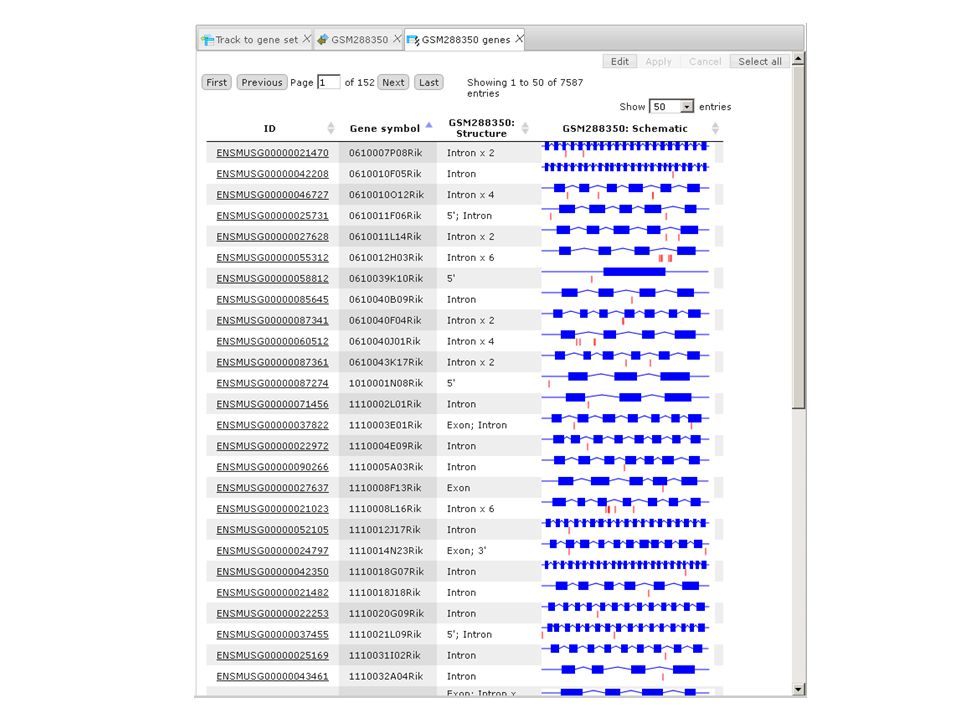
Genotyping Affected genes Affected genes http://cloud-biotech.com/bioumlweb/ #de=data/Collaboration/Dr.Archakov/Data/alignment/ Ap1.bam-CleanedAlignment/Genotyping2/tmp/Raw-affected-annotated

59
Genotyping: potential lose of function 118 genes118 genes have mutations that potentially affect function Mutation in the exon of MAP4K3

60
Gene ontology classification Full table

61
Genome browser Example of deletion and insertion presentation in genome browserdeletioninsertion

62
Ribosome profiling

64
Live demonstration

Similar presentations