Download presentation
Presentation is loading. Please wait.

1
Polymerase Chain Reaction
What is PCR History of PCR How PCR works Optimizing PCR Fidelity, errors & cloning PCR primer design Application of PCR

2
Polymorphism among 32 wheat samples revealed by AFLP

3
Characteristics of AFLP
dominant marker. DNA variation is detected by presence/absence of DNA bands due to: a) presence/absence of restriction sites b) additional bases (insertion) between two restriction sites are too large
 presence/absence of restriction sites. b) additional bases (insertion) between two restriction sites are too large.")
4
Advantages higher reproducibility compared to RAPD.
highly polymorphic.

5
Think … …

6
PCR Requirements Magnesium chloride: .5-2.5mM Buffer: pH 8.3-8.8
dNTPs: µM Primers: µM DNA Polymerase: units Target DNA: 1 µg

7
Can I PCR Amplify RNA? Not directly — the DNA polymerase requires a DNA template and will not copy RNA. mRNA can first be copied into cDNA using reverse transcriptase. cDNA is a template for PCR —it need not be double-stranded.

8
Cloning PCR Products Products should be ligatable into blunt-ended restriction enzyme site. Lower than expected efficiency. Products are not truly blunt-ended. Taq polymerase adds a single nontemplated base (usually A) to the 3´ end: NNNNNNN…NNNNNNNA ANNNNNNN…NNNNNNN
 to the 3´ end: NNNNNNN…NNNNNNNA. ANNNNNNN…NNNNNNN.")
9
Cloning PCR-generated Fragments
Generation of compatible ends. T/A cloning vectors

10
PCR Mutagenesis Add restriction sites for cloning Generate deletional mutants Generate single point mutations

11
THE PROBLEM QUANTITATION OF mRNA northern blotting
ribonuclease protection assay in situ hybridization PCR most sensitive can discriminate closely related mRNAs technically simple but difficult to get truly quantitative results using conventional PCR Northern blotting and RPAS are the gold standards, since no amplification is involved. In situ hybridization is qualitative rather than quantitative. Techniques such as Northern blotting and ribonuclease protection assays (RPAs) work very well, but require more RNA than is sometimes available. PCR methods are particularly valuable when amounts of RNA are low, since the fact that PCR involves an amplification step means that it is more sensitive. However, traditional PCR is only semi-quantitative at best, in part because of the insensitivity of ethidium bromide (however, there are more sensitive ways to detect the product) and, in part, as we shall discuss later, because of the difficulties of observing the reaction during the truly linear part of the amplification process. Various competitive PCR protocols have been designed to overcome this problem but they tend to be cumbersome. Real-time PCR has been developed so that more accurate results can be obtained. An additional advantage of real-time PCR is the relative ease and convenience of use compared to some of these older methods (as long as one has access to a suitable real-time PCR machine).
 work very well, but require more RNA than is sometimes available. PCR methods are particularly valuable when amounts of RNA are low, since the fact that PCR involves an amplification step means that it is more sensitive. However, traditional PCR is only semi-quantitative at best, in part because of the insensitivity of ethidium bromide (however, there are more sensitive ways to detect the product) and, in part, as we shall discuss later, because of the difficulties of observing the reaction during the truly linear part of the amplification process. Various competitive PCR protocols have been designed to overcome this problem but they tend to be cumbersome. Real-time PCR has been developed so that more accurate results can be obtained. An additional advantage of real-time PCR is the relative ease and convenience of use compared to some of these older methods (as long as one has access to a suitable real-time PCR machine).")
12
Protocol RT-PCR Isolate RNA (total or polyA selected)
Use RNA to generate cDNA (reverse transcriptase) Use cDNA from RT reaction as template for PCR amplification.
 Use cDNA from RT reaction as template for PCR amplification.")
13
RT-PCR vs “Real-time” RT-PCR
RT-PCR is endpoint analysis “Real-time” (fluorescent; quantitative) PCR is continual analysis
 PCR is continual analysis.")
14
Application of “Real-time” PCR
Expression analysis (RNA) Identification/quantification of single base pair polymorphisms (DNA) Genotyping (DNA) Exposure monitoring RNA) Clinical Screening Cancer (DNA) Pathogen detection (DNA/RNA)
 Identification/quantification of single base pair polymorphisms (DNA) Genotyping (DNA) Exposure monitoring RNA) Clinical Screening. Cancer (DNA) Pathogen detection (DNA/RNA)")
15
Considerations for Fluorescent “Real-time” RT-PCR
Primer sets Chosing target sequence Checking primer sets Chosing fluorescent label Quantification Relative quantification Absolute quantification Standard curves

16
Basics of real time RT-PCR Abundance of mRNA
Central hypothesis: Changes in gene expression reflect changes in cell function Nearly all physiological changes in animals are accompanied by changes in gene expression Immune response Toxic response Pharmacological response

17
Outline Basics of PCR and real time PCR Experimental Design
Data Analysis Application to Drug Development and Design

18
Polymerase Chain Reaction
In vitro method for the amplification of short (up to ~5000bp) pieces of DNA Relies on a thermostable form of DNA polymerase Thermus aquaticus
 pieces of DNA. Relies on a thermostable form of DNA polymerase. Thermus aquaticus.")
19
Polymerase Chain Reaction
Required reagents: Template DNA Primers* DNA polymerase dNTPs* Thermocycler

20
Polymerase Chain Reaction
Proper controls Water blank Concentration curve Internal standard control Genomic DNA contaminant control

21
Internal standard Gene of interest

22
Polymerase Chain Reaction
Advantages Sensitive Versatile – easy to test new genes Primers are inexpensive Reliable Much more than microarrays for individual transcripts Standardized competitor templates or standard curves Allow comparison between expts Internal standards Addresses variation in tissue starting amounts or loading errors

23
Polymerase Chain Reaction
Disadvantages Optimizations required Annealing temperature Number of cycles Small order of magnitiude sensitivity for detection

24
AFLP Amplified Fragment Length Polymorphism

27
Principles of Real-Time Quantitative PCR Techniques
SYBR Green I technique: SYBR Green I fluorescence is enormously increased upon binding to double-stranded DNA. During the extension phase, more and more SYBR Green I will bind to the PCR product, resulting in an increased fluorescence. Consequently, during each subsequent PCR cycle more fluorescence signal will be detected. Hydrolysis probe technique: The hydrolysis probe is conjugated with a quencher fluorochrome, which absorbs the fluorescence of the reporter fluorochrome as long as the probe is intact. However, upon amplification of the target sequence, the hydrolysis probe is displaced and subsequently hydrolyzed by the Taq polymerase. This results in the separation of the reporter and quencher fluorochrome and consequently the fluorescence of the reporter fluorochrome becomes detectable. During each consecutive PCR cycle this fluorescence will further increase because of the progressive and exponential accumulation of free reporter fluorochromes. Hybridization probes technique: In this technique one probe is labelled with a donor fluorochrome at the 3’ end and a second –adjacent- probe is labelled with an acceptor fluorochrome. When the two fluorochromes are in close vicinity (1–5 nucleotides apart), the emitted light of the donor fluorochrome will excite the acceptor fluorochrome (FRET). This results in the emission of fluorescence, which subsequently can be detected during the annealing phase and first part of the extension phase of the PCR reaction. After each subsequent PCR cycle more hybridization probes can anneal, resulting in higher fluorescence signals.
, the emitted light of the donor fluorochrome will excite the acceptor fluorochrome (FRET). This results in the emission of fluorescence, which subsequently can be detected during the annealing phase and first part of the extension phase of the PCR reaction. After each subsequent PCR cycle more hybridization probes can anneal, resulting in higher fluorescence signals.")
28
What do mRNA levels tell us?
DNAmRNAprotein Reflect level of gene expression Information about cell response Protein production (not always)
")
29
quantitative mRNA/DNA analysis
Direct -Northern blotting -In situ hybridization PCR amplification -Regular RT-PCR -Real time PCR (Microarrays) There two general ways to analyse mRNA. Either directly like in in situ hybridiszation or throug PCR magnification. Which include regular PCR and real time PCR. nöfn
 There two general ways to analyse mRNA. Either directly like in in situ hybridiszation or throug PCR magnification. Which include regular PCR and real time PCR. nöfn.")
30
Why isn´t this good enough?
This slide shows the conventional PCR based testing formats. mRNA extraction, DNA amplification and then analysis by either by gel electrophorezis which can possable be followed by a Northern blot and Enzyme linked hybridisation which is not as common. Why isn´t this good enough?

31
What’s Wrong With Agarose Gels?
* Low sensitivity * Low resolution * Non-automated * Size-based discrimination only * Results are not expressed as numbers based on personal evaluation Ethidium bromide staining is not very quantitative End point analysis huglægt?? ABI: Real-Time PCR vs Traditional PCR (www)
")
32
Different concentrations give similar endpoint results!
Endpoint analysis exponential increase Different concentrations give similar endpoint results!

33
Real-time Principles based on the detection and quantitation of a fluorescent reporter In stead of measuring the endpoint we focus on the first significant increase in the amount of PCR product. The time of the increase correlates inversely to the initial amount of DNA template

34
Polymerization Q R R = Reporter Q = Quencher 3 Probe Forward Primer
5 Reverse R = Reporter Q = Quencher

35
For Real Time PCR we need a a specific probe with a fluorescent reporter.
Q R

36
When in close contact with the reporter,
the quencer absobes its emission.

37
Strand Displacement 3 Q 5 R

38
Cleavage 3 Q R 5

39
Polymerization Completed
3 Q R 5

40
Van der Velden. Leukemia 2003 (www)
")
41
* nonspecific binding is a disadvantage
SYBR Green (double-stranded DNA binding dye) * emits a strong fluorescent signal upon binding to double-stranded DNA * nonspecific binding is a disadvantage * requires extensive optimisation longer amplicons create a stronger signal It´s cheap
 * emits a strong fluorescent signal upon binding to double-stranded DNA. * nonspecific binding is a disadvantage. * requires extensive optimisation. longer amplicons create a stronger signal. It´s cheap.")
42
SYBR® Green I Chemistry
Polymerization Forward Primer 5' 3' 5' 5' 3' 5' Reverse Primer Polymerization completed 5' 3' 5' 5' 3' 5'

43
Real-time PCR advantages
* not influenced by non-specific amplification * amplification can be monitored real-time * no post-PCR processing of products (high throughput, low contamination risk) * requirement of 1000-fold less RNA than conventional assays (3 picogram = one genome equivalent) * most specific, sensitive and reproducible
 * requirement of 1000-fold less RNA than conventional assays. (3 picogram = one genome equivalent) * most specific, sensitive and reproducible.")
44
Housekeeping gene Knowing the amount of mRNA in one sample from one specific gene does not tell us alot You don´t know the total amount of mRNA in your sample You also dont know how much the mRNA level has changed compared to other mRNA levels Example: mRNA levels increase 2x after induction It is possable that all genexpression in the cell has increased We have to compare the expression of our gene to another gene which expression is normally constant, a housekeeping gene

45
Real-time PCR advantages
* not influenced by non-specific amplification * amplification can be monitored real-time * no post-PCR processing of products (high throughput, low contamination risk) * ultra-rapid cycling (30 minutes to 2 hours) * wider dynamic range of up to 1010-fold * requirement of 1000-fold less RNA than conventional assays (3 picogram = one genome equivalent) * detection is capable down to a 2-fold change * confirmation of specific amplification by melting curve analysis * most specific, sensitive and reproducible * not much more expensive than conventional PCR (except equipment cost)
 * ultra-rapid cycling (30 minutes to 2 hours) * wider dynamic range of up to 1010-fold. * requirement of 1000-fold less RNA than conventional assays. (3 picogram = one genome equivalent) * detection is capable down to a 2-fold change. * confirmation of specific amplification by melting curve analysis. * most specific, sensitive and reproducible. * not much more expensive than conventional PCR. (except equipment cost)")
46
Real-time PCR disadvantages
* setting up requires high technical skill and support * high equipment cost * * * * intra- and inter-assay variation * RNA lability * DNA contamination (in mRNA analysis)
")
47
Principles of Real-Time Quantitative PCR Techniques
SYBR Green I technique: SYBR Green I fluorescence is enormously increased upon binding to double-stranded DNA. During the extension phase, more and more SYBR Green I will bind to the PCR product, resulting in an increased fluorescence. Consequently, during each subsequent PCR cycle more fluorescence signal will be detected. Hydrolysis probe technique: The hydrolysis probe is conjugated with a quencher fluorochrome, which absorbs the fluorescence of the reporter fluorochrome as long as the probe is intact. However, upon amplification of the target sequence, the hydrolysis probe is displaced and subsequently hydrolyzed by the Taq polymerase. This results in the separation of the reporter and quencher fluorochrome and consequently the fluorescence of the reporter fluorochrome becomes detectable. During each consecutive PCR cycle this fluorescence will further increase because of the progressive and exponential accumulation of free reporter fluorochromes. Hybridization probes technique: In this technique one probe is labelled with a donor fluorochrome at the 3’ end and a second –adjacent- probe is labelled with an acceptor fluorochrome. When the two fluorochromes are in close vicinity (1–5 nucleotides apart), the emitted light of the donor fluorochrome will excite the acceptor fluorochrome (FRET). This results in the emission of fluorescence, which subsequently can be detected during the annealing phase and first part of the extension phase of the PCR reaction. After each subsequent PCR cycle more hybridization probes can anneal, resulting in higher fluorescence signals.
, the emitted light of the donor fluorochrome will excite the acceptor fluorochrome (FRET). This results in the emission of fluorescence, which subsequently can be detected during the annealing phase and first part of the extension phase of the PCR reaction. After each subsequent PCR cycle more hybridization probes can anneal, resulting in higher fluorescence signals.")
48
Schematic diagram comparing three different fluorescence-monitoring systems for DNA amplification. System A uses dsDNA-specific dyes (F) such as SYBR"Green I, which increase in fluorescence when bound to accumulating amplification product. System B uses dual-labelled probes and depends on the 5'-exonuclease activity of the polymerase to separate donor (D) and acceptor (A) by hydrolysis. Donor fluorescence is increased by removing acceptor quenching. System C depends on the independent hybridization of adjacent donor (D) and acceptor (A) probes. Their approximation increases resonance energy transfer from the donor to the acceptor. Other symbols are "hv" for excitation light and "x" for a 3'-phosphate.
 such as SYBR Green I, which increase in fluorescence when bound to accumulating amplification product. System B uses dual-labelled probes and depends on the 5 -exonuclease activity of the polymerase to separate donor (D) and acceptor (A) by hydrolysis. Donor fluorescence is increased by removing acceptor quenching. System C depends on the independent hybridization of adjacent donor (D) and acceptor (A) probes. Their approximation increases resonance energy transfer from the donor to the acceptor. Other symbols are hv for excitation light and x for a 3 -phosphate..")
49
TaqMan Probes FRET = Förster/fluorescence resonance energy transfer &
DNA Polymerase 5' exonuclease activity * Tm value 100 C higher than primers * runs of identical nucleotides (no consecutive Gs) * G+C content 30-80% * more Cs than Gs * no G at the 5' end
 * G+C content 30-80% * more Cs than Gs. * no G at the 5 end.")
50
FRET = Förster/fluorescence resonance energy transfer
ABI: Real-Time PCR vs Traditional PCR (www)
")
51
DNA Polymerase 5' Exonuclease Activity
Mocellin et al. Trends Mol Med 2003 (www)
")
52
The TaqMan 5’ Exonuclease Assay
In addition to two conventional PCR primers, P1 and P2, which are specific for the target sequence, a third primer, P3, is designed to bind specifically to a site on the target sequence downstream of the P1 binding site. P3 is labelled with two fluorophores, a reporter dye (R) is attached at the 5 end, and a quencher dye (D), which has a different emission wavelength to the reporter dye, is attached at its 3 end. Because its 3 end is blocked, primer P3 cannot by itself prime any new DNA synthesis. During the PCR reaction, Taq DNA polymerase synthesizes a new DNA strand primed by P1 and as the enzyme approaches P3, its exonuclease activity processively degrades the P3 primer from its 5 end. The end result is that the nascent DNA strand extends beyond the P3 binding site and the reporter and quencher dyes are no longer bound to the same molecule. As the reporter dye is no longer in close proximity to the quencher, the resulting increase in reporter emission intensity is easily detected. Human Molecular Genetics 2. NCBI Books (www)
 is attached at the 5 end, and a quencher dye (D), which has a different emission wavelength to the reporter dye, is attached at its 3 end. Because its 3 end is blocked, primer P3 cannot by itself prime any new DNA synthesis. During the PCR reaction, Taq DNA polymerase synthesizes a new DNA strand primed by P1 and as the enzyme approaches P3, its 5 3 exonuclease activity processively degrades the P3 primer from its 5 end. The end result is that the nascent DNA strand extends beyond the P3 binding site and the reporter and quencher dyes are no longer bound to the same molecule. As the reporter dye is no longer in close proximity to the quencher, the resulting increase in reporter emission intensity is easily detected. Human Molecular Genetics 2. NCBI Books (www)")
53
TaqMan Primers * equal Tm (58-600 C) * 15-30 bases in length
* G+C content 30-80% * no runs of four or more Gs (any nucleotide) * no more than two G+C at the 3’ end * no G at the 5' end * amplicon size bp (max 400) * span exon-exon junctions in cDNA
 * no more than two G+C at the 3’ end. * no G at the 5 end. * amplicon size bp (max 400) * span exon-exon junctions in cDNA.")
54
SYBR Green (double-stranded DNA binding dye) * emits a strong fluorescent signal upon binding to double-stranded DNA * nonspecific binding is a disadvantage * requires extensive optimization * requires melting point curve determination * longer amplicons create a stronger signal * may be multiplexed when coupled with melting curve analysis

55
Fluoresces when bound to dsDNA

56
SYBR Green (1) At the beginning of amplification, the reaction mixture contains the denatured DNA, the primers, and the dye. The unbound dye molecules weakly fluoresce, producing a minimal background fluorescence signal which is subtracted during computer analysis. (2) After annealing of the primers, a few dye molecules can bind to the double strand. DNA binding results in a dramatic increase of the SYBR Green I molecules to emit light upon excitation. (3) During elongation, more and more dye molecules bind to the newly synthesized DNA. If the reaction is monitored continuously, an increase in fluorescence is viewed in real-time. Upon denaturation of the DNA for the next heating cycle, the dye molecules are released and the fluorescence signal falls.
 At the beginning of amplification, the reaction mixture contains the denatured DNA, the primers, and the dye. The unbound dye molecules weakly fluoresce, producing a minimal background fluorescence signal which is subtracted during computer analysis. (2) After annealing of the primers, a few dye molecules can bind to the double strand. DNA binding results in a dramatic increase of the SYBR Green I molecules to emit light upon excitation. (3) During elongation, more and more dye molecules bind to the newly synthesized DNA. If the reaction is monitored continuously, an increase in fluorescence is viewed in real-time. Upon denaturation of the DNA for the next heating cycle, the dye molecules are released and the fluorescence signal falls.")
57
When to Choose SYBR Green
* Assays that do not require specificity of probe based assays. Detection of 1000s of molecules * General screening of transcripts prior to moving to probe based assays * When the PCR system is fully optimized -no primer dimers or non-specific amplicons, e.g. from genomic DNA

58
When Not to Choose SYBR Green
* Allelic discrimination assays (not an absolute one) * Multiplex reactions (not an absolute one) * Amplification of rare transcripts * Low level pathogen detection
 * Multiplex reactions (not an absolute one) * Amplification of rare transcripts. * Low level pathogen detection.")
59
Real-Time Principles Three general methods for the quantitative detection: 1. Hydrolysis probes (TaqMan, Beacons, Scorpions) 2. Hybridization probes (Light Cycler) 3. DNA-binding agents (SYBR Green)
![]()
60
Mocellin et al. Trends Mol Med 2003 (www)
Molecular Beacons Mocellin et al. Trends Mol Med 2003 (www)
")
61
Real-Time Principles Three general methods for the quantitative detection: 1. Hydrolysis probes (TaqMan, Beacons, Scorpions) 2. Hybridization probes (Light Cycler) 3. DNA-binding agents (SYBR Green)
![]()
62
Scorpions Bustin SA. J Mol Endocrinol 2002 (www)
")
63
Scorpions Bustin SA. J Mol Endocrinol 2002 (www)
")
64
* it is the parameter used for quantitation
Threshold Cycle * threshold cycle or the CT value is the cycle at which a significant increase in DRn is first detected * it is the parameter used for quantitation * CT value of 40 or more means no amplification and cannot be included in the calculations

65
What is CT? The Amplification Plot contains valuable information for the quantitative measurement of DNA or RNA. The Threshold line is the level of detection or the point at which a reaction reaches a fluorescent intensity above background. The threshold line is set in the exponential phase of the amplification for the most accurate reading. The cycle at which the sample reaches this level is called the Cycle Threshold, CT. These two values are very important for data analysis using the 5’ nuclease assay.

67
Albumin (ALB) gene dosage by real-time PCR
Laurendeau et al. Clin Chem 1999 (www)
")
68
DRn * Rn+ is the Rn value of a reaction containing all components (the sample of interest); Rn- is the Rn value detected in NTC (baseline value) * DRn is the difference between Rn+ and Rn-. It is an indicator of the magnitude of the signal generated by the PCR * DRn is plotted against cycle numbers to produce the amplification curves and gives the CT value

69
What is DRn? +Rn Rn Rn -Rn CT cycle number Sample 10 20 30 40
10 20 30 40 cycle number Rn CT Threshold Rn Sample No Template Control -Rn +Rn

70
What is DRn? (www)
")
71
Endogenous/Internal Control (Normalization)
* usually an abundantly and constantly expressed housekeeping gene * most commonly used ones are the least reliable ones * best to run a validity test for the selected endogenous control * combination may/should be used

72
Endogenous Control Selection
Sabek et al. Transplantation 2002 (www)
")
73
Approximation vs Pfaffl method (Efficiency Determination)
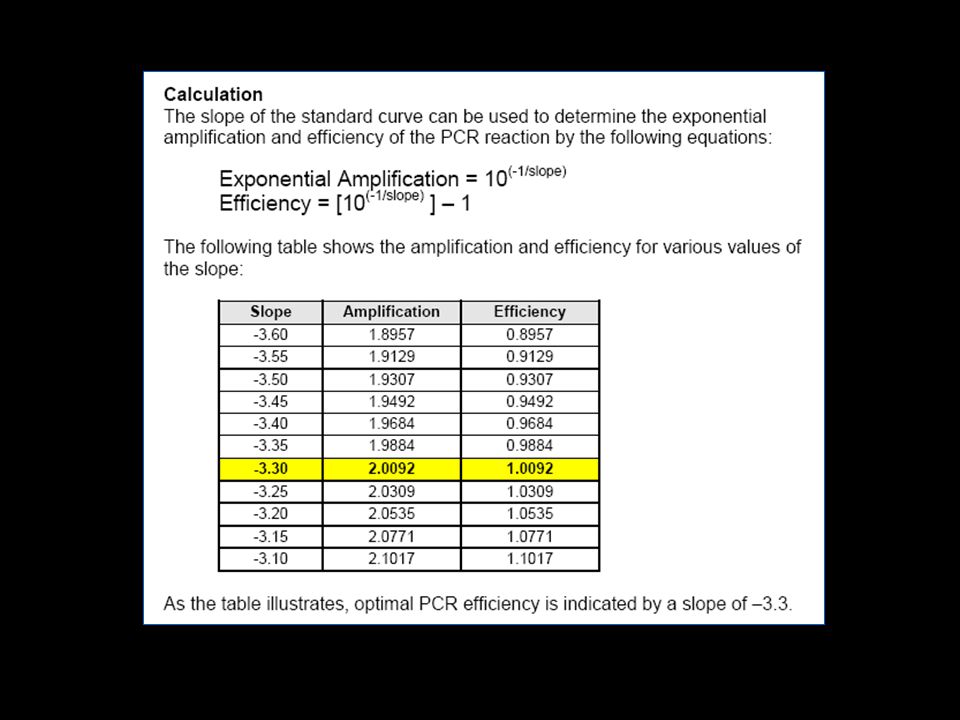
The slope of the log-linear phase is a reflection of the amplification efficiency The efficiency of the reaction can be calculated by the following equation: Eff=10(-1/slope) –1. The efficiency of the PCR should be % (ideal slope = 3.3) A number of variables can affect the efficiency of the PCR. These factors can include length of the amplicon, secondary structure, and primer design, to name a few Approximation vs Pfaffl method (Efficiency Determination)
 –1. The efficiency of the PCR should be % (ideal slope = 3.3) A number of variables can affect the efficiency of the PCR. These factors can include length of the amplicon, secondary structure, and primer design, to name a few. Approximation vs Pfaffl method. (Efficiency Determination)")
75
Using the PCR Equation Xn X0 Xn = X0(1 + E)n
Xn = PCR product after cycle n X0 = initial copy number E = amplification efficiency n = cycle number E IS MAXIMALLY 1.0, BUT USUALLY AROUND 0.9. X0 cycle number

76
Effect of Amplification Efficiency
Xn = X0(1+E)n Case 1: E = Case 2: E = 0.8 Xn = 100 (1+0.9)30 Xn = 100 (1+0.8)30 Xn = 2.3 x 1010 Xn = 4.6 x 109 CALCULATE EFFICIENCIES FROM THE SLOPE OF YOUR DOSE RESPONSE CURVES Result A difference of 0.1 in amplification efficiencies created a five-fold difference in the final ratio of PCR products after 30 cycles
n. Case 1: E = 0.9 Case 2: E = 0.8. Xn = 100 (1+0.9)30. Xn = 100 (1+0.8)30. Xn = 2.3 x Xn = 4.6 x 109. CALCULATE EFFICIENCIES FROM THE SLOPE OF YOUR DOSE RESPONSE CURVES. Result. A difference of 0.1 in amplification. efficiencies created a five-fold difference in the. final ratio of PCR products after 30 cycles.")
77
Determination of real-time PCR efficiencies of reference gene (Gst), target gene 1 (TyrA) and target gene 2 (PyrB). CP cycles versus cDNA (reverse transcribed total RNA) concentration input were plotted to calculate the slope (mean ± SD; n = 3). The corresponding real-time PCR efficiencies were calculated according to the equation: E = 10[–1/slope]
 concentration input were plotted to calculate the slope (mean ± SD; n = 3). The corresponding real-time PCR efficiencies were calculated according to the equation: E = 10[–1/slope].")
78
If the CT values for each of the dilutions are plotted against concentrations, the result should be a linear graph with a high correlation coefficient (> 0.99). The slope of this graph is also a measure of efficiency, and can be readily used to calculate efficiency - this is done by most software (iCycler, for example).
..")
79
Nigel Walker, NIEHS (www)
")
80
Nigel Walker, NIEHS (www)
")
81
Assay Validation * Test primer pairs in all combinations with the probe with a known template (plasmid clone, sDNA, RNA) * Use standard assay conditions: nM primers; 100 nM probe, 3 mM MgCl2 * Choose the primer pair that gives the highest DRn and the lowest CT * Make a dilution of a template, either sDNA, sRNA or total RNA for a standard curve * Correlation coefficient of the standard curve > 0.99? * If the slope of the standard curve of the best primer pair is around -3.5 increase the MgCl2 to 5 mM * If the slope is higher than -3.6, change primers * An ideal assay will have a slope of -3.3

82
One-Step or Two-Step PCR
* one-step real-time RT-PCR performs reverse transcription and PCR in a single buffer system and in one tube * in two-step RT-PCR, these two steps are performed separately in different tubes

83
The really big deal Isolation of DNA polymerase from Thermus
aquaticus (Mullis and Faloona 1987) • T. aquaticus is a hot springs bacterium • Therefore, has thermally stable DNA polymerase (e.g. not killed by heat)
 • T. aquaticus is a hot springs bacterium. • Therefore, has thermally stable DNA. polymerase (e.g. not killed by heat)")
84
Importance of T. aquaticus
Extension at 72 C: Most polymerases are active at 45 C or below (e.g. normal body/environmental temps)-temps below normal annealing temps-results in nonspecific binding of primers 2. From Bacterium that lives in near boiling water: Most polymerase inactivated by high temperatures-initially had to add new polymerase each cycle
-temps below. normal annealing temps-results in nonspecific. binding of primers. 2. From Bacterium that lives in near boiling. water: Most polymerase inactivated by high. temperatures-initially had to add new. polymerase each cycle.")
85
Colony PCR CPSC265 Class 8

86
Cloning Cloning is the way in which we can take a single molecule, and make lots of bacterial cells that contain an identical molecule. These cells are clones, hence the name This used to be the only way to amplify DNA. It is still by far the most accurate.

88
Plasmid vectors – circular, autonomous bacterial DNA

89
The vector is made with a “T” overhang

90
Taq polymerase leaves an “A” overhang
Taq is the thermostable DNA polymerase from Thermus aquaticus we used for PCR. When Taq synthesizes a new strand, it always puts an extra “A” at the end This can be useful, but note: other polymerases do not do this, they leave “blunt” ends. Only Taq polymerase leaves ‘A’ overhangs. ‘Blunt’ end vectors do not work with Taq, we need a ‘T’ overhang.

92
DNA ligase Repairs gaps in the sugar-phosphate backbone of DNA
Creates phosphodiester bonds Does not do anything with the bases

93
Transformation of bacteria
Two main methods for transformation Chemical / Heat Shock As done in last practical, this method gets DNA into the cell by making them porous using CaCl2 and a 42 C heat treatment Electroporation Makes cells porous using high-voltage electricity

94
Imperfect science Most of the plasmid / insert combinations will not ligate Most of the bacteria will not be transformed We only need one molecule to get into one bacterium to make one colony.

95
PCR from clones Often clones will religate containing any old DNA (eg primer dimers).. The DNA can go in in either orientation We can use the PCR to tell which colonies have the insert we want, and which orientation it is in.

96
Some population genetics software
Microsatellite toolkit: Excel plug-in for creating Arlequin, FSTAT and Genepop files. Microchecker: Estimate null allele frequency. Adjust allele frequencies. Arlequin: HW equilibrium, Linkage Disequilibrium, Fst, exact test of differentiation, Amova, Mantel test FSTAT: Allelic richness, Fst per locus (to check contribution of each locus to observed pattern of differentiation) Structure, BAPS: Population structuring, population assignment. Migrate: Estimates of effective population size and migration rates Bottleneck: Check for very recent population bottlenecks
 Structure, BAPS: Population structuring, population assignment. Migrate: Estimates of effective population size and migration rates Bottleneck: Check for very recent population bottlenecks")
Similar presentations






>")






 Analysis of DNA (Sequencing) Chemical Synthesis.>")







>")


