Download presentation
Presentation is loading. Please wait.

1
p53 has a key role in integrating the cellular responses (pink boxes) to different types of stress (blue boxes). Activation of p53 can result in a number of cellular responses, and it is possible that different responses are induced by different stress signals. There is evidence that p53 can play a part in determining which response is induced through differential activation of target-gene expression. Although the importance of these responses to tumour suppression is clear, previously unanticipated contributions of these responses to other aspects of human health and disease are being uncovered. The role of p53 in tumour suppression, development and ageing is likely to depend on which cellular response is activated and on the context in which the activation occurs.

2
p53 is at the centre of a complex web of biological interactions that translates stress signals into cell cycle arrest or apoptosis26. Upstream signalling to p53 increases its level and activates its function as a transcription factor in response to a wide variety of stresses, whereas downstream components execute the appropriate cellular response. The principal sensors seem to be MDM2 and MDM4 and their interaction with p53. In non-stressed conditions these proteins bind p53, ubiquitylate it and target it for degradation by the proteasome. In stressed conditions the function of the MDM2–MDM4 complex is blocked by phosphorylation, protein-binding events and/or enhanced degradation141. Hence, phosphorylation of MDM4 is essential for the p53 response to ionizing radiation, and the response to oncogene activation depends on the binding of ARF to MDM2. Many p53-activating small molecules function by causing the release of ribosomal proteins from the nucleolus to the nucleoplasm, where they bind to MDM2 and MDM4 and inhibit their function. Molecules that activate wild-type p53 in tumours by disrupting MDM2 activity can compensate for any missing upstream components of the p53 pathway, for example the loss of ARF expression that is frequent in cancer cells142. However, defective downstream p53 signalling might substantially decrease their effectiveness. Therefore, the ability to identify tumours in which downstream p53 signalling is unaffected is important. The development of strategies to ensure that the desired p53 response is initiated when it is reactivated might be necessary and could require the judicious use of drug combinations. 53BP1, p53 binding protein 1; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related; BAI1, brain-specific angiogenesis inhibitor 1; BAX, BCL2-associated X protein; BBC3, BCL2 binding component 3 (also known as PUMA); DR, death receptor; GADD45, growth arrest and DNA-damage-inducible 45; KILLER, p53-regulated DNA damage-inducible cell death receptor (also known as TNFRSF10B); LRDD, leucine-rich repeats and death domain containing; miRNA, microRNA; PMAIP1, phorbol-12-myristate-13-acetate-induced protein 1 (also known as NOXA); RPRM, reprimo; RRM2B, ribonucleotide reductase M2 B; ST13, suppression of tumorigenicity 13 (also known as p48); TP53I3, tumour protein p53 inducible protein 3; THBS1, thrombospondin 1; UV, ultraviolet.
; DR, death receptor; GADD45, growth arrest and DNA-damage-inducible 45; KILLER, p53-regulated DNA damage-inducible cell death receptor (also known as TNFRSF10B); LRDD, leucine-rich repeats and death domain containing; miRNA, microRNA; PMAIP1, phorbol-12-myristate-13-acetate-induced protein 1 (also known as NOXA); RPRM, reprimo; RRM2B, ribonucleotide reductase M2 B; ST13, suppression of tumorigenicity 13 (also known as p48); TP53I3, tumour protein p53 inducible protein 3; THBS1, thrombospondin 1; UV, ultraviolet..")
3
The p53–MDM2 feedback loop is the 'heart' of the p53 pathway
The p53–MDM2 feedback loop is the 'heart' of the p53 pathway. Under normal conditions, it maintains constantly low steady-state p53 levels and activity. Various stress signals (only a representative subset of p53-activating signals is depicted), related in many ways to carcinogenesis, impinge on this central loop to release p53 from MDM2-mediated inhibition. This increases p53 protein levels and activity, inducing various phenotypic changes. Many p53-activating signals are closely interrelated, as exemplified here for oncogenes, the effect of which on p53 is partly due to their propensity to induce DNA replication stress. The downstream effects of p53 are largely due to its ability to transactivate and repress various subsets of target genes; however, at least in the case of apoptosis, protein–protein interactions (primarily with Bcl2 family members) also have an important role. It is generally believed that the nature of the phenotypic response to p53 activation is, at least partially, proportionate to the amplitude, duration and nature of the activating signal. Severe stress induces more extreme, usually irreversible, responses, namely apoptosis and senescence, whereas milder stress leads to a transient growth arrest coupled with an attempt to deal with the cause of stress and repair the damage caused by it. Recent evidence indicates that p53 also has an important role in enabling the cell to adjust its metabolism in response to mild normal physiological fluctuations, including those in glucose and other nutrient levels, oxygen availability and reactive oxygen species levels (see the Review by Vousden and Ryan131, also in this issue).
, related in many ways to carcinogenesis, impinge on this central loop to release p53 from MDM2-mediated inhibition. This increases p53 protein levels and activity, inducing various phenotypic changes. Many p53-activating signals are closely interrelated, as exemplified here for oncogenes, the effect of which on p53 is partly due to their propensity to induce DNA replication stress. The downstream effects of p53 are largely due to its ability to transactivate and repress various subsets of target genes; however, at least in the case of apoptosis, protein–protein interactions (primarily with Bcl2 family members) also have an important role. It is generally believed that the nature of the phenotypic response to p53 activation is, at least partially, proportionate to the amplitude, duration and nature of the activating signal. Severe stress induces more extreme, usually irreversible, responses, namely apoptosis and senescence, whereas milder stress leads to a transient growth arrest coupled with an attempt to deal with the cause of stress and repair the damage caused by it. Recent evidence indicates that p53 also has an important role in enabling the cell to adjust its metabolism in response to mild normal physiological fluctuations, including those in glucose and other nutrient levels, oxygen availability and reactive oxygen species levels (see the Review by Vousden and Ryan131, also in this issue).")
4
In normal cells, p53 increases transcription of MDM2 over basal levels
In normal cells, p53 increases transcription of MDM2 over basal levels. MDM2 also inhibits p53 function by modulating its transcriptional activity by preventing its interaction with the general transcription machinery28. MDM2 also forms a heterodimeric complex with MDM4 that promotes the degradation of p53 (Ref. 92). ARF adds another level of control to the system by inhibiting MDM2 function143, the expression of which is in turn also repressed by p53 (Ref. 144). MDM2, as well as promoting p53 degradation, ubiquitylates (Ub) MDM4 (Refs 94) and promotes the degradation of the Mdm2–MDM4 complex in response to ionizing radiation. This is mediated at least in part through ATM- and CHK2- dependent phosphorylation of MDM4, which inhibits binding to proteins and the deubiquitylating enzyme USP7 (also known as herpes virus-associated ubiquitin-specific protease (HAUSP)), thus permitting its proteasomal destruction141, 145, 146. These cellular mechanisms result in subtle control of p53 levels. ATM, ataxia telangiectasia mutated; DUB, deubiquitylase.
. ARF adds another level of control to the system by inhibiting MDM2 function143, the expression of which is in turn also repressed by p53 (Ref. 144). MDM2, as well as promoting p53 degradation, ubiquitylates (Ub) MDM4 (Refs 94) and promotes the degradation of the Mdm2–MDM4 complex in response to ionizing radiation. This is mediated at least in part through ATM- and CHK2- dependent phosphorylation of MDM4, which inhibits binding to proteins and the deubiquitylating enzyme USP7 (also known as herpes virus-associated ubiquitin-specific protease (HAUSP)), thus permitting its proteasomal destruction141, 145, 146. These cellular mechanisms result in subtle control of p53 levels. ATM, ataxia telangiectasia mutated; DUB, deubiquitylase.")
5
Under unstressed conditions, MDM2 and p53 associate through their amino-termini (step 1), which leads to the acidic domain of MDM2 making contact with the Box IV–V region in p53 (step 2). This allows the subsequent ubiquitylation of p53 (Refs 132, 133, 134, 135) (step 3). DNA damage stimulates ataxia–telangiectasia mutated (ATM)-dependent and ataxia–telangiectasia and Rad3-related (ATR)-dependent phosphorylation of MDM2, leading to MDM2 degradation51, 52, 53. In addition, ATM and ATR directly phosphorylate serine 15 of p53 (Refs 136, 137, 138, 139), protein kinase CK1 phosphorylates threonine 18 (using phosphorylated S15 as a priming site)140, 141 and S20 is phosphorylated by CHK2 (which is activated by ATM)142, 143, 144. Many other modifications of p53 are dependent either indirectly upon ATM or occur sequentially following the phosphorylation of S15 (Refs 141, 145, 146, 147). Biochemical analyses and studies using cultured cells indicate that the phosphorylation of these p53 sites stimulates the recruitment of key transcriptional proteins, such as p300 and CBP148, 149, 150, 151, 152, 153, 154, 155, leading to the acetylation of several key lysine residues in the carboxy-terminus of p53 that are normally targets for ubiquitylation: this process is thought to help stabilize p53 (Refs 147, 156). Phosphorylation of T18 and S20 also inhibit the association of p53 with MDM2 (Refs 140, 141, 157, 158, 159, 160). Several stresses target the crucial acidic domain of MDM2: DNA damage-mediated hypophosphorylation inhibits MDM2-mediated p53 degradation161, 162, and interaction with the ARF tumour suppressor inhibits MDM2 function (see Fig. 4 and Refs 163, 164). Ac, acetyl; DNA-BD, DNA-binding domain; M, methyl; NES, nuclear export signal; NLS, nuclear localization signal; P, phosphate; p53BD, p53-binding domain; Pro, proline-rich region; TAD, transactivation domain.
 (step 3). DNA damage stimulates ataxia–telangiectasia mutated (ATM)-dependent and ataxia–telangiectasia and Rad3-related (ATR)-dependent phosphorylation of MDM2, leading to MDM2 degradation51, 52, 53. In addition, ATM and ATR directly phosphorylate serine 15 of p53 (Refs 136, 137, 138, 139), protein kinase CK1 phosphorylates threonine 18 (using phosphorylated S15 as a priming site)140, 141 and S20 is phosphorylated by CHK2 (which is activated by ATM)142, 143, 144. Many other modifications of p53 are dependent either indirectly upon ATM or occur sequentially following the phosphorylation of S15 (Refs 141, 145, 146, 147). Biochemical analyses and studies using cultured cells indicate that the phosphorylation of these p53 sites stimulates the recruitment of key transcriptional proteins, such as p300 and CBP148, 149, 150, 151, 152, 153, 154, 155, leading to the acetylation of several key lysine residues in the carboxy-terminus of p53 that are normally targets for ubiquitylation: this process is thought to help stabilize p53 (Refs 147, 156). Phosphorylation of T18 and S20 also inhibit the association of p53 with MDM2 (Refs 140, 141, 157, 158, 159, 160). Several stresses target the crucial acidic domain of MDM2: DNA damage-mediated hypophosphorylation inhibits MDM2-mediated p53 degradation161, 162, and interaction with the ARF tumour suppressor inhibits MDM2 function (see Fig. 4 and Refs 163, 164). Ac, acetyl; DNA-BD, DNA-binding domain; M, methyl; NES, nuclear export signal; NLS, nuclear localization signal; P, phosphate; p53BD, p53-binding domain; Pro, proline-rich region; TAD, transactivation domain..")
6
Oncogene activation is thought to lead to the induction of ARF and consequent activation of p53 and tumour suppression90, 91. Oncogene-induced DNA damage has been proposed as an alternative mechanism through which p53 tumour suppressor function is alerted to the presence of aberrant proliferative factors72, 75. In both cases, there is a selective pressure for the inactivation of checkpoint components to allow developing tumours to progress to malignancy.

7
The CDKN2A locus encodes ARF in an overlapping reading frame with the tumour suppressor INK4A (also known as p16), and ARF is normally expressed at low levels in cells. Hyperproliferative signals lead to the increased expression of ARF, which inhibits MDM2 by blocking its E3 ubiquitin ligase activity58 (mechanism 1), uncoupling the p53–MDM2 interaction57 (mechanism 2) and sequestering MDM2 in the nucleolus, thereby segregating it from nucleoplasmic p53 (Refs 60, 61, 62) (mechanism 3).
, uncoupling the p53–MDM2 interaction57 (mechanism 2) and sequestering MDM2 in the nucleolus, thereby segregating it from nucleoplasmic p53 (Refs 60, 61, 62) (mechanism 3)..")
8
a | A model for p53 activation based on a persistent oncogenic signal
a | A model for p53 activation based on a persistent oncogenic signal. The threshold for activation is triggered by signals persisting beyond a specified time point, so the duration of the signal determines activation. Note that the intensity of the oncogenic insult does not influence when activation occurs (left panel compared with right panel). b | A model for activation based on the strength of the oncogenic signal. The threshold for activation is fixed at a specific level, making signal intensity the crucial determinant of activation (left panel). Low-level, persistent oncogenic signals evade this means of activation and undetectably drive tumorigenesis (right panel).
. b | A model for activation based on the strength of the oncogenic signal. The threshold for activation is fixed at a specific level, making signal intensity the crucial determinant of activation (left panel). Low-level, persistent oncogenic signals evade this means of activation and undetectably drive tumorigenesis (right panel).")
9
Regulation of p53, a stress-regulated transcription factor that co-ordinately induces or represses sets of gene products in response to changes in the cellular microenvironment. P53 co-operates with the transcriptional activator p300 to induce sets of gene products implicated in growth control (left panel). P53 co-operates with de-acetylases to repress gene expression of proteins implicated in proliferation and survival (right panel). P53 is often mutated in human cancers leading to a protein that can not respond to signalling cues and that can not regulate gene products that orchestrate the stress response. The mutation in p53 unfolds the protein, stabilizes it in the nucleus where it can be bound by molecular chaperones, and creates a protein that often has an oncogenic gain-of-function property.
. P53 co-operates with de-acetylases to repress gene expression of proteins implicated in proliferation and survival (right panel). P53 is often mutated in human cancers leading to a protein that can not respond to signalling cues and that can not regulate gene products that orchestrate the stress response. The mutation in p53 unfolds the protein, stabilizes it in the nucleus where it can be bound by molecular chaperones, and creates a protein that often has an oncogenic gain-of-function property..")
10
miR-34 is a direct transcriptional target of p53, which in turn downregulates genes required for proliferation and survival. Along with other p53 targets, such as p21 and BAX, miR-34-family miRNAs promote growth arrest and cell death in response to cancer related stress. ATM, ataxia talangiectasia mutated; ATR, ataxia telengiectasia and RAD3-related; CDK, cyclin-dependent kinase; CHK, checkpoint kinase

11
INACTIVATION OF THE p53 PATHWAY
Dominant-negative missense p53 gene mutation Upstream tumour suppressor gene mutation (ATM) Oncogene amplification (MDM2) Viral oncogene amplification (HPV E6)
 Oncogene amplification (MDM2) Viral oncogene amplification (HPV E6)")
13
Structure and expression of p53 family members
Structure and expression of p53 family members. (a) Structure of p53, p63 and p73 transcription units. Numbered boxes indicate exons, and black shading denotes untranslated sequences. The approximate regions encoding the transactivation (TA) domain (light blue), N-specific region (green), DNA-binding domain (red), oligomerization domain (yellow), sterile alpha motif (SAM, grey), and transactivational inhibitory domain (TID, orange) are indicated. Distinct transcription start sites are indicated by arrows. N-terminal alternative splicing for p53 and p73 are indicated by dotted lines, and C-terminal splicing events for all p53 family members are indicated by solid lines and Greek letter designation. (b) Protein domains of p53 family members. All three family members share a homologous DNA-binding domain and oligomerization domain (oligo). The TA domain is shared by p53, TAp63, and TAp73 isoforms. TAp63 /TAp73 isoforms most closely resemble p53. N-terminally truncated N isoforms possess unique N-terminal sequences. Alpha isoforms of p63 and p73 possess a C-terminal SAM domain followed by a transactivational inhibitory domain (TID). Other isoforms of p53, p63 and p73 are not shown.
 Structure of p53, p63 and p73 transcription units. Numbered boxes indicate exons, and black shading denotes untranslated sequences. The approximate regions encoding the transactivation (TA) domain (light blue), N-specific region (green), DNA-binding domain (red), oligomerization domain (yellow), sterile alpha motif (SAM, grey), and transactivational inhibitory domain (TID, orange) are indicated. Distinct transcription start sites are indicated by arrows. N-terminal alternative splicing for p53 and p73 are indicated by dotted lines, and C-terminal splicing events for all p53 family members are indicated by solid lines and Greek letter designation. (b) Protein domains of p53 family members. All three family members share a homologous DNA-binding domain and oligomerization domain (oligo). The TA domain is shared by p53, TAp63, and TAp73 isoforms. TAp63 /TAp73 isoforms most closely resemble p53. N-terminally truncated N isoforms possess unique N-terminal sequences. Alpha isoforms of p63 and p73 possess a C-terminal SAM domain followed by a transactivational inhibitory domain (TID). Other isoforms of p53, p63 and p73 are not shown.")
14
Structure and expression of p53 family members
Structure and expression of p53 family members. (a) Structure of p53, p63 and p73 transcription units. Numbered boxes indicate exons, and black shading denotes untranslated sequences. The approximate regions encoding the transactivation (TA) domain (light blue), N-specific region (green), DNA-binding domain (red), oligomerization domain (yellow), sterile alpha motif (SAM, grey), and transactivational inhibitory domain (TID, orange) are indicated. Distinct transcription start sites are indicated by arrows. N-terminal alternative splicing for p53 and p73 are indicated by dotted lines, and C-terminal splicing events for all p53 family members are indicated by solid lines and Greek letter designation. (b) Protein domains of p53 family members. All three family members share a homologous DNA-binding domain and oligomerization domain (oligo). The TA domain is shared by p53, TAp63, and TAp73 isoforms. TAp63 /TAp73 isoforms most closely resemble p53. N-terminally truncated N isoforms possess unique N-terminal sequences. Alpha isoforms of p63 and p73 possess a C-terminal SAM domain followed by a transactivational inhibitory domain (TID). Other isoforms of p53, p63 and p73 are not shown.
 Structure of p53, p63 and p73 transcription units. Numbered boxes indicate exons, and black shading denotes untranslated sequences. The approximate regions encoding the transactivation (TA) domain (light blue), N-specific region (green), DNA-binding domain (red), oligomerization domain (yellow), sterile alpha motif (SAM, grey), and transactivational inhibitory domain (TID, orange) are indicated. Distinct transcription start sites are indicated by arrows. N-terminal alternative splicing for p53 and p73 are indicated by dotted lines, and C-terminal splicing events for all p53 family members are indicated by solid lines and Greek letter designation. (b) Protein domains of p53 family members. All three family members share a homologous DNA-binding domain and oligomerization domain (oligo). The TA domain is shared by p53, TAp63, and TAp73 isoforms. TAp63 /TAp73 isoforms most closely resemble p53. N-terminally truncated N isoforms possess unique N-terminal sequences. Alpha isoforms of p63 and p73 possess a C-terminal SAM domain followed by a transactivational inhibitory domain (TID). Other isoforms of p53, p63 and p73 are not shown.")
15
Figure 3. Model for p63 as a tumor suppressor
Figure 3. Model for p63 as a tumor suppressor. (A) Down regulation or loss of TAp63 and/ or overexpression of ΔNp63 leads to inhibition of the functions of TAp63, p53, and TAp73 leading to the development of an invasive and metastatic tumor. (B) Mutant p53 binds to TAp63 and TAp73 inhibiting their function leading to the development of an invasive and metastatic tumor.
 Down regulation or loss of TAp63 and/ or overexpression of ΔNp63 leads to inhibition of the functions of TAp63, p53, and TAp73 leading to the development of an invasive and metastatic tumor. (B) Mutant p53 binds to TAp63 and TAp73 inhibiting their function leading to the development of an invasive and metastatic tumor.")
16
Pathways involving the p53 family functional network in squamous cell carcinoma. DNA damage and oncogenic stress (through p14arf) trigger p53 activation, leading to selective pressure for its mutation or loss to avoid apoptosis. Similarly, DNA damage and potentially oncogenic stress induce TAp73 expression and activation. TAp73 activity can be suppressed through overexpression of Np63 or certain missense p53 mutants (mtp53). Alternatively, apoptosis can be averted in Np63 -negative tumors through overexpression of bcl-2, which inhibits the proapoptotic function of TAp73 target genes.
 trigger p53 activation, leading to selective pressure for its mutation or loss to avoid apoptosis. Similarly, DNA damage and potentially oncogenic stress induce TAp73 expression and activation. TAp73 activity can be suppressed through overexpression of Np63 or certain missense p53 mutants (mtp53). Alternatively, apoptosis can be averted in Np63 -negative tumors through overexpression of bcl-2, which inhibits the proapoptotic function of TAp73 target genes..")
17
p63/p73 pathway mediates cisplatin sensitivity in squamous carcinoma cells. In proliferating tumor cells, Np63 inhibits the proapoptotic transcriptional activity of TAp73 through both direct physical interaction and through direct binding to the promoters of TAp73 target genes. Cisplatin treatment induces Np63 downregulation and TAp73 stabilization, thereby activating the TAp73-dependent apoptotic program. Phosphorylation of TAp73 by c-Abl and potentially other kinases is important for its activation following DNA damage. Phosphorylation may also contribute to downregulation of Np63 in this context. Note that proteins are shown schematically as dimers but in fact are thought to bind DNA as tetramers.

18
| Most inactivating mutations of p53 occur in the DNA-binding domain (DBD), whereas the amino-terminal transactivation domain (TA) is relatively free of point mutations. Mutations in yellow affect DNA contacts, those in light blue cause local distortions and those in dark blue cause global denaturation. b | Therapeutics that disrupt MDM2 binding to the TA, for example nutlin8, are designed to reactivate wild-type p53 by displacing MDM2, which is overexpressed in many p53 wild-type tumours. The crystal structure of nutlin8 (cyan) in complex with MDM2 shows how it mimics the three key residues from p53 (orange) that are involved in the interaction. The small molecule PhiKan083 has also been designed to specifically interact with the p53-Y220C mutant. This demonstrates the possibility of therapeutically targeting tumours with different inactivating mutations of p53. Parts c and d of the figure show the cleft generated on the surface of p53 by the missense mutation (c), where PhiKan083 (Ref. 69) has also been shown to bind using crystallography (d). These two therapeutic strategies are designed to reactivate p53 but differ in that the first method reactivates wild-type p53 and the second reactivates mutant p53. PR, proline-rich domain; Reg, carboxy-terminal regulatory domain; Tet, tetramerization domain.
 in complex with MDM2 shows how it mimics the three key residues from p53 (orange) that are involved in the interaction. The small molecule PhiKan083 has also been designed to specifically interact with the p53-Y220C mutant. This demonstrates the possibility of therapeutically targeting tumours with different inactivating mutations of p53. Parts c and d of the figure show the cleft generated on the surface of p53 by the missense mutation (c), where PhiKan083 (Ref. 69) has also been shown to bind using crystallography (d). These two therapeutic strategies are designed to reactivate p53 but differ in that the first method reactivates wild-type p53 and the second reactivates mutant p53. PR, proline-rich domain; Reg, carboxy-terminal regulatory domain; Tet, tetramerization domain..")
19
421 421 240 240 1620 1620 (A) wildtype or (B) structural mutants
DNA contact mutants (B) structural mutants 421 421 240 240 1620 1620 1620 421 240 1620 421 240
 structural mutants")
20
The inner circle (shaded blue) represents oncogenic phenotypes associated with the activities of mutant p53 proteins. The outer circle depicts key mechanistic properties of p53 mutants that underlie the phenotypes listed in the inner circle. Note that each of the phenotypic effects can be attributed to almost each of the mechanistic properties; hence the inner blue circle can be freely rotated. ATM, ataxia-telangiectasia mutated; NF-B, nuclear factor-B; VDR, vitamin D receptor.

21
Figure 1. Mutant p53 is stabilized by unknown mechanisms in tumors
Figure 1. Mutant p53 is stabilized by unknown mechanisms in tumors. Sufficient mutant p53 is then available to bind and inactivate the functions of wild type p53, p63 and p73.

23
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING p53 MUTATIONS Gene therapy with wt p53

24
pRb E1A p53 E1B 55kDa E1B E1B 19kDa E1A E1B ADENOVIRUS
CELL CYCLE ARREST E1A ADENOVIRUS p53 APOPTOSIS DEGRADATION E1B 55kDa E1B E1B 19kDa APOPTOSIS E1A E1B rAd-p53 REPLACED BY DA p53wt

25
DRAWBACKS IN THE USE OF ADENOVIRAL VECTORS FOR GENE THERAPY
Transient gene expression (14-21d) Prevalenceof neutralizing antibodies Access to tumor cells
 Prevalenceof neutralizing antibodies. Access to tumor cells.")
26
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING p53 MUTATIONS Gene therapy with wt p53 Selectively replicating oncolytic viruses (Onyx-015)
")
27
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING p53 MUTATIONS Gene therapy with wt p53 Selectively replicating oncolytic viruses (Onyx-015) Direct activation of p53 target gene products (p21waf1; Bax) pharmacological rescue of mutant p53 protein: Hsp90 inhibitors others
 Direct activation of p53 target gene products (p21waf1; Bax) pharmacological rescue of mutant p53 protein: Hsp90 inhibitors. others.")
28
pRb E1A p53 E1B 55kDa E1B E1B 19kDa pRb E1A E1B DEGRADATION ADENOVIRUS
APOPTOSIS ONYX-015 E1A pRb E1B

29
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING p53 MUTATIONS Gene therapy with wt p53 Selectively replicating oncolytic viruses (Onyx-015) Direct activation of p53 target gene products (p21waf1; Bax)
 Direct activation of p53 target gene products (p21waf1; Bax)")
30
Correcting mutant p53 to perform wild-type functions
Correcting mutant p53 to perform wild-type functions. Specific p53 mutants maybe altered to perform wild type-mediated growth inhibition through exposure to the activating molecules (Section 3 and subsections within). Haupt S Semin Cancer Biol 2004
. Haupt S Semin Cancer Biol")
31
(p53 reactivation and induction of massive apoptosis)
PRIMA-1 (p53 reactivation and induction of massive apoptosis)
")
32
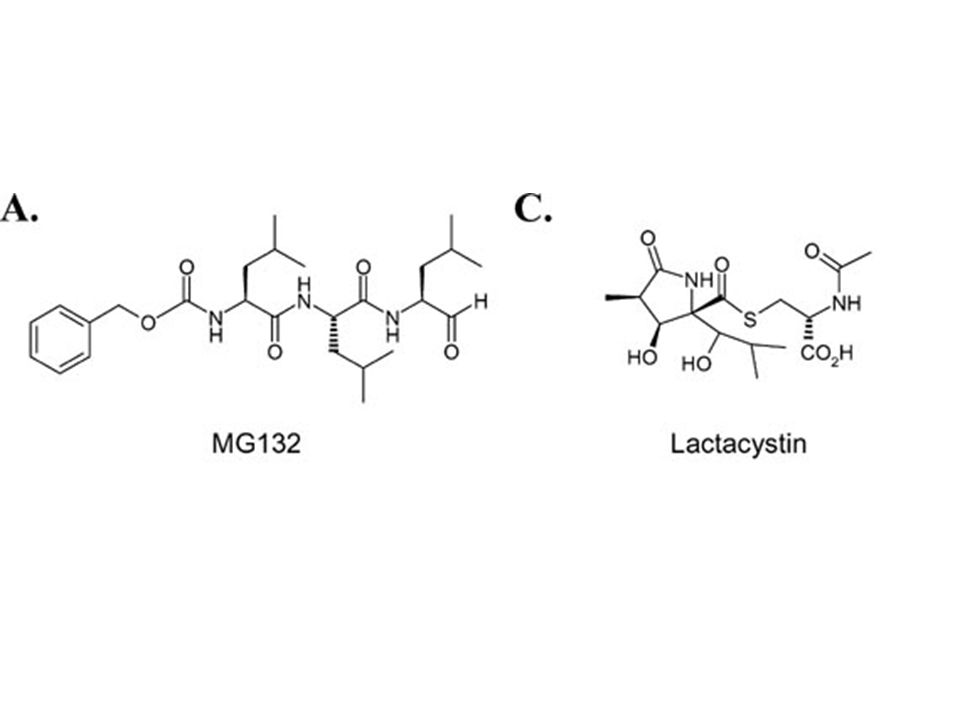
CP does not bind the core domain of recombinant wt or mutant p53 in vitro. It has no effect on the interaction between p53 and Mdm2. Hypothesis: CP is proposed to interact with newly synthetized p53 in vivo, blocking its ubiquitination and degradation

33
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING wildtype p53 p53 response activation Block of E6 expression Inhibition of Mdm2 expression Inhibition of Mdm2 function

34
Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).
![Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).](http://slideplayer.com/slide/6066660/18/images/34/Structural+formula+of+PRIMA-1+%282%2C2-bis%28hydroxy-methyl%29-1-azabicyclo%5B2%2C2%2C2%5Doctan-3-one%29..jpg "Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).")
35
Regulation of p53 by MDM2. p53 and MDM2 form an auto-regulatory feedback loop. p53 stimulates the expression of MDM2; MDM2 inhibits p53 activity because it blocks its transcriptional activity, favours its nuclear export and stimulates its degradation. Different cellular signals, such as DNA-damage or oncogene activation, induce p53 activation. DNA damage favours p53 phosphorylation, preventing its association with MDM2. Activated oncogenes activate the ARF protein, which prevents the MDM2-mediated degradation of p53. Similarly, inhibitors of the p53–MDM2 interaction should activate p53 tumour-suppressor activity in tumour cells that express wild-type p53. These compounds, because they bind to MDM2, could also affect the p53-independent activities of MDM2.

36
Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).
![Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).](http://slideplayer.com/slide/6066660/18/images/36/Structural+formula+of+PRIMA-1+%282%2C2-bis%28hydroxy-methyl%29-1-azabicyclo%5B2%2C2%2C2%5Doctan-3-one%29..jpg "Structural formula of PRIMA-1 (2,2-bis(hydroxy-methyl)-1-azabicyclo[2,2,2]octan-3-one).")
37
FIGURE 1. Mdm2/Mdmx heterodimers are more effective p53 ubiquitin ligases than Mdm2 homodimers
Wade, M. et al. Mol Cancer Res 2009;7:1-11

39
RITA (Reactivation of p53 and Induction of Tumor cell Apoptosis)
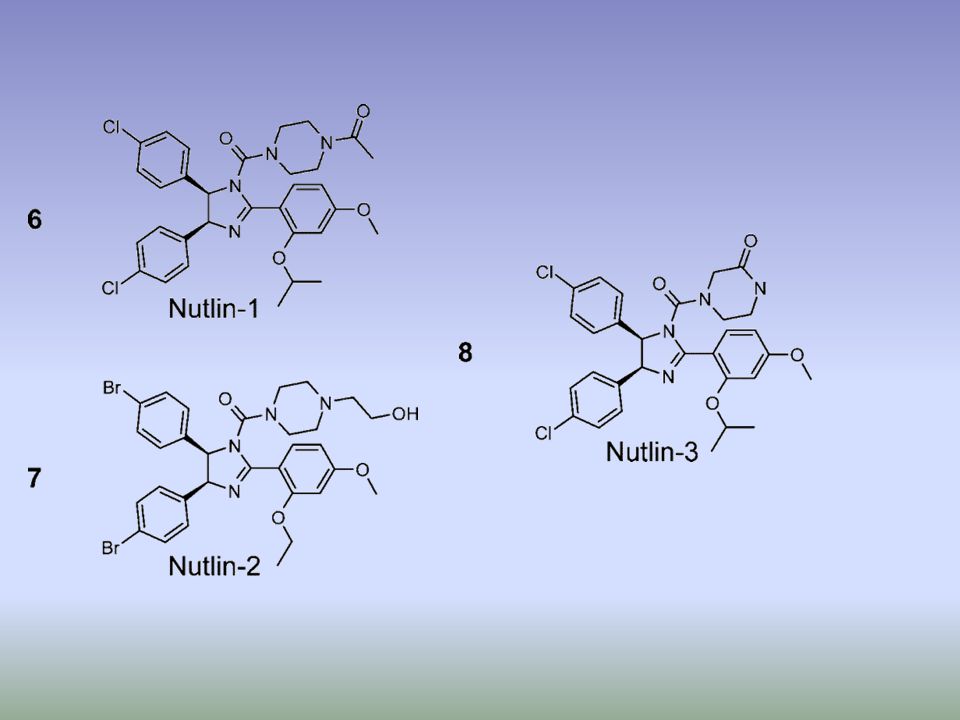
. b | Structure of Nutlin (left) and RITA (right), the first small-molecule inhibitors of the p53–MDM2 interaction73, 75. c | Structure of the Nutlin–p53 complex19. Left: the sides of p53-binding groove in MDM2 (red) is limited by two -helices and a short -sheet while the bottom is formed by two shorter -helices perpendicular to the pocket sides. The amino-terminal domain of p53 (green) is stabilized in -helical conformation upon binding to MDM2 due to a network of hydrogen bonds trapping three crucial residues of p53, Phe19, Trp23 and Leu26. These crucial p53 residues point towards the bottom of the groove. Right: Nutlin prevents interaction between p53 and MDM2 by mimicking the conformation of Phe19, Trp23 and Leu26 of p53 to block the MDM2 p53-interacting domain73. MDM2, double minute 2. RITA (Reactivation of p53 and Induction of Tumor cell Apoptosis)
 and RITA (right), the first small-molecule inhibitors of the p53–MDM2 interaction73, 75. c | Structure of the Nutlin–p53 complex19. Left: the sides of p53-binding groove in MDM2 (red) is limited by two -helices and a short -sheet while the bottom is formed by two shorter -helices perpendicular to the pocket sides. The amino-terminal domain of p53 (green) is stabilized in -helical conformation upon binding to MDM2 due to a network of hydrogen bonds trapping three crucial residues of p53, Phe19, Trp23 and Leu26. These crucial p53 residues point towards the bottom of the groove. Right: Nutlin prevents interaction between p53 and MDM2 by mimicking the conformation of Phe19, Trp23 and Leu26 of p53 to block the MDM2 p53-interacting domain73. MDM2, double minute 2. RITA (Reactivation of p53 and Induction of Tumor cell Apoptosis)")
41
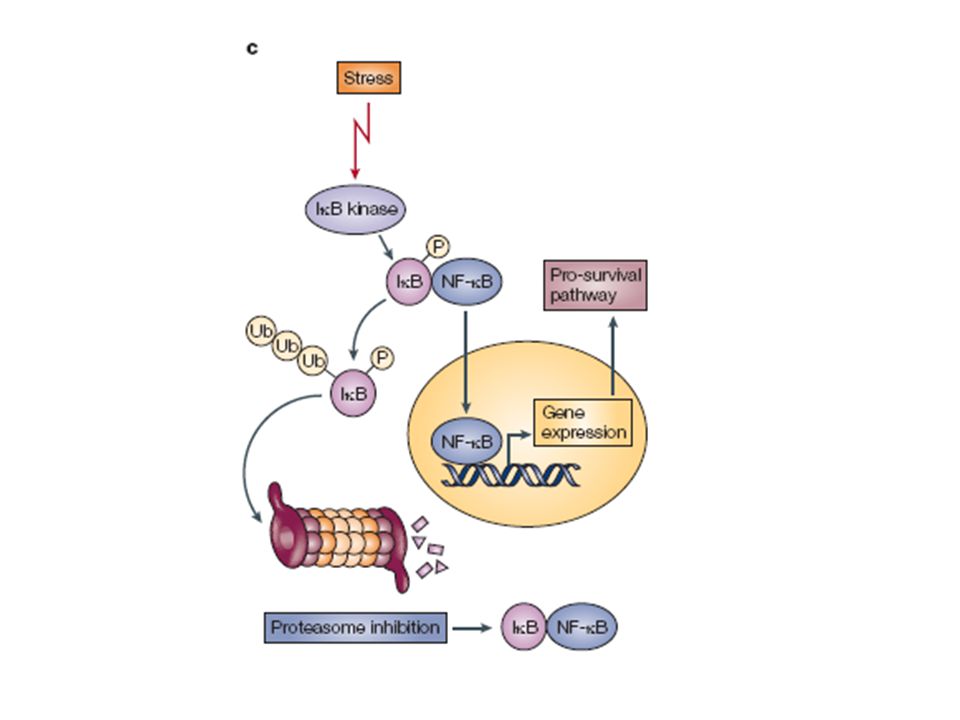
Promotion of wt p53 activities through protection from Mdm2-mediated inhibition. Interruption of the p53–Mdm2 interaction can be achieved by treatment with Nutlins, syc-7 and chalcones leading to activation of p53-mediated growth inhibition (Section 2.1). Down regulation of Mdm2 can be achieved through the introduction of anti-sense mRNA directed toward Mdm2. The drug Cp appears to activate wt p53 by preventing p53 ubiquitination and possibly through the induction of DNA damage (Section ). The proteasome inhibitor bortezomib stabilizes p53 by preventing its degradation mediated by Mdm2, possibly by inhibiting p53 ubiquitination ( Section 2.4). Green arrows denote activating pathways while red bars denote inhibition.
. Down regulation of Mdm2 can be achieved through the introduction of anti-sense mRNA directed toward Mdm2. The drug Cp appears to activate wt p53 by preventing p53 ubiquitination and possibly through the induction of DNA damage (Section ). The proteasome inhibitor bortezomib stabilizes p53 by preventing its degradation mediated by Mdm2, possibly by inhibiting p53 ubiquitination ( Section 2.4). Green arrows denote activating pathways while red bars denote inhibition..")
42
Proteasome structure. A three-dimensional representation of the proteasome multi-enzyme complex. This is composed of the 20S complex, which comprises - and -subunits, and two 19S regulatory complexes. Together with ATP, these form the 26S proteasome.

43
20

45
BORTEZOMIB (VELCADE®)
The effects of proteasome inhibition on the stability Figure 3 The structure of the dipeptidyl boronic acid proteasome inhibitor bortezomib. BORTEZOMIB (VELCADE®)
")
46
Figure 4 Cross-sectional view of the bortezomib binding site
in the proteasome. Bortezomib interacts with a threonine residue located on the b subunit that confers chymotryptic proteolytic activity.

49
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING wildtype p53 p53 response activation Block of E6 expression Inhibition of Mdm2 expression Inhibition of Mdm2 function

50
THERAPEUTIC STRATEGIES DIRECTED AT p53
TUMORS BEARING wildtype p53 p53 response activation Block of E6 expression Inhibition of Mdm2 expression Inhibition of Mdm2 function Block of p53 nuclear trafficking (leptomycin B)
")
52
Leptomycin B (LMB) is an unsaturated branched-chain fatty acid and can undergo a Michael-type addition by Cysteine-529 on the exportin CRM1. This covalent modification inhibits binding between CRM1 and the nuclear export signal (NES)-containing cargo. Nuclear transport as a target for cell growth, Pages Tweeny R. Kau and Pamela A. Silver

53
Some of the points at which p53 can affect metabolic pathways
Some of the points at which p53 can affect metabolic pathways. This is a new and rapidly moving area of research, and the influence of p53 on metabolism is likely to be much broader than illustrated here. In response to nutrient stress, p53 can become activated by AMP kinase (AMPK), promoting cell survival through an activation of the cyclin-dependent kinase inhibitor p21. Other functions of p53 include regulating respiration, through the action of SCO2, or in decreasing the levels of reactive oxygen species (ROS), through the actions of TIGAR (Tp53-inducible glycolysis and apoptosis regulator) or sestrins.
, promoting cell survival through an activation of the cyclin-dependent kinase inhibitor p21. Other functions of p53 include regulating respiration, through the action of SCO2, or in decreasing the levels of reactive oxygen species (ROS), through the actions of TIGAR (Tp53-inducible glycolysis and apoptosis regulator) or sestrins.")
54
p53 functions in the response to both the constitutive stress that is encountered during normal growth and development, and to the acute stress signals that would be associated with oncogenic progression and other types of trauma. In this model, p53 responds to conditions of low or constitutive stress to play an important part in decreasing oxidative damage, and provides repair functions to mend low levels of DNA damage. These activities of p53 contribute to the survival and health of the cell as well as to the prevention of the acquisition of tumorigenic mutations, and might contribute to overall longevity and normal development. By contrast, acute stress that results in a more robust induction of p53 leads to the activation of apoptotic cell death and thereby the elimination of the damaged cell. This function removes cells that have acquired oncogenic alterations, and can contribute to neural tube closure during development, but carries accompanying detrimental effects of stress-induced toxicity, such as radiation sickness, neurodegenerative disease and premature ageing.

55
A recent study from Evan and colleagues33 provides evidence that the immediate p53 response to DNA damage is detrimental and not necessary for tumour suppression. The absence of p53 immediately after DNA damage protects animals from radiation sickness, but does not prevent repair of the DNA damage. Persistent absence of p53 results in tumour development, as expected. However, even transient restoration of p53 activity after the resolution of the initial DNA damage can inhibit tumour development without the deleterious responses, such as widespread apoptosis in lymphoid organs and intestinal epithelium, that occur following the irradiation of mice with fully active p53. These results suggest that the induction of p53 in response to signals that persist beyond DNA damage, such as activated oncogenes, is the key to tumour suppression.

56
pifithrin-

Similar presentations






 Cancer Res. 54:4855 50%>")
 associated anogenital disease and potential for vaccination Peter L. Stern Journal of Clinical Virology, 2005.>")




 has a definite role in cancer.>")
 mechanisms.>")










