Download presentation
Presentation is loading. Please wait.

1
Drug Stability

2
Drug stability It refers to the capacity of a drug substance or product to remain within established specifications of identity, strength, quality and purity in a specified period of time. Stability is officially defined as the time lapse during which the drug product retains the same properties and characteristics that it possessed at the time of manufacture. The stability of a product is expressed as the expiry period or technically as shelf-life.

3
Objectives of Stability Study
1- Provide an evidence on how the quality of a drug substance or drug product varies with time under the influence of a variety of environmental factors such as • temperature, • humidity, • and light 2- Establish a: • re-test period for the drug substance or a • shelf life for the drug product and • recommended storage conditions

4
To gather information during preformulation stage to produce a stable product.
- To determine maximum expiration date. - To gate on idea of storage conditions. - To determine the packaging components. The retest period of pharmaceuticals. Transport conditions

5
Purposes of stability studies
The purposes of stability studies are to predict and confirm product shelf-life under the climatic conditions expected during trade storage, shipping, house storage, and use.

6
● Chemical degradation of active drug may reduce the quality of therapeutic indices like 5-fluorouracil, carbamazepine etc have very small therapeutic range, slight degradation of drug may produce sub-therapeutic concentration. ● After degradation, a drug may produce more toxic product (s) which may be more toxic than the parent product. ● Instability of drug product reduce bioavailability. This may be caused by physical or chemical instability. ● Instability of a product may change the physical appearance of the product
 which may be more toxic than the parent product. ● Instability of drug product reduce bioavailability. This may be caused by physical or chemical instability. ● Instability of a product may change the physical appearance of the product.")
7
Factors affecting drug stability
Storage time Storage conditions Type of dosage form Container and closure system

8
1- Environmental factors
- Temperature Light - Oxygen Moisture - Carbon dioxide 2- Drugs or excipients in the dosage form Particle size of drug pH of the vehicle 3- Microbial contamination 4- Trace metal contamination 5- Leaching from containers

9
Chemical stability implies: - The lack of any decomposition in the chemical moiety that is incorporated in the formulation as the drug, preservatives or any other excipients This decomposition may influence the physical and chemical stability of the drug

10
Microbiological stability implies that:
- The formulation has not suffered from any microbiological attack and is meeting the standards with respect to lack of contamination/sterility.

11
Physical changes • Appearance • Melting point • Clarity and color of solution • Crystal modification (Polymorphism) • Particle size Chemical changes • Increase in Degradation products • Decrease of Assay Microbial changes • Growth of microorganism
 • Particle size Chemical changes • Increase in Degradation products • Decrease of Assay Microbial changes • Growth of microorganism")
12
Packaging And Stability :
The immediate container and closure are particularly important in affecting product stability. They play an important role in the product shelf-life. They may accelerate degradation reactions, be an additive to or an absorbent of the drug substance, or be ineffective in protecting the contents from environmental conditions.

13
Glass - Glass is resistant to chemical and physical change and is the most commonly used materials
Limitations overcomes 1. Its alkaline surface may raise the pH of the pharmaceutical and induce chemical reaction. 2- Ionic radicals in the drug may precipitate insoluble crystals from the glass such as barium sulfate. 3- Permits the transmission of light which may accelerate physical and chemical reactions in the drug. use of Borosilicate glass which contains fewer reactive alkali ions than the other 3 types of USP-recognized glass Treatment the glass with heat as well as the use of buffers. Amber colored glass reducing light-induced reactions.

14
Plastics The problems with plastic are:
1. Migration of the drug through the plastic into the environment. 2. Transfer of environmental moisture, oxygen, and other elements into the pharmaceutical product. 3. Leaching of container ingredients into the drug. 4.Adsorption or absorption of the active drug or excipients by the plastic.

15
Metals Various alloys and aluminum tubes may be utilized as containers for emulsions, ointments, creams and pastes. Limitation: They may cause corrosion and precipitation in the drug product especially with products at extreme pH values or those containing metallic ions. Overcome: Coating the tubes with polymers may reduce these tendencies

16
Rubber Rubber also has the problems of extraction of drug ingredients and leaching of container ingredients. The use of neoprene, butyl or natural rubber, in combination with certain epoxy, Teflon, or vanish coating, substantially reduces drug-container interaction. The pretreatment of rubber vial stoppers and closures with water and steam removes surface blooms and also reduces potential leaching that might affect chemical analysis, toxicity, or pyrogenicity of the drug formulation

17
Stability studies at different stages
1. Stress- and accelerated Testing with drug substances 2. Stability on pre-formulation batches 3. Stress testing on scale-up Batches 4. Accelerated and long term testing for registration 5. On-going Stability testing 6. Follow-up Stabilities

18
Stability testing There shall be a written testing program designed to assess the stability characteristics of drug products. The results of such stability shall be used in determining appropriate storage conditions and expiration dates. The written program shall be followed and shall include: 1) Sample size and test intervals based on statistical criteria for each attribute examined to assure valid estimates of stability. 2) Storage conditions for samples retained for testing. 3) Reliable, meaningful, and specific test methods. 4) Testing of the drug product in the same container-closure system as that in which the drug product is marketed. 5) Testing of drug products for reconstitution at the time of dispensing (as directed in the labeling) as well as after they are reconstituted.
 Sample size and test intervals based on statistical criteria for each attribute examined to assure valid estimates of stability. 2) Storage conditions for samples retained for testing. 3) Reliable, meaningful, and specific test methods. 4) Testing of the drug product in the same container-closure system as that in which the drug product is marketed. 5) Testing of drug products for reconstitution at the time of dispensing (as directed in the labeling) as well as after they are reconstituted.")
20
An adequate number of batches of each drug product shall be tested to determine an appropriate expiration date and a record of such data shall be maintained. For homeopathic drug products, the requirements of this section are as follows: (1) There shall be a written assessment of stability based at least on testing or examination of the drug product for compatibility of the ingredients, and based on marketing experience with the drug product to indicate that there is no degradation of the product for the normal or expected period of use. (2) Evaluation of stability shall be based on the same container-closure system in which the drug product is being marketed.
 There shall be a written assessment of stability based at least on testing or examination of the drug product for compatibility of the ingredients, and based on marketing experience with the drug product to indicate that there is no degradation of the product for the normal or expected period of use. (2) Evaluation of stability shall be based on the same container-closure. system in which the drug product is being marketed.")
21
● Before commencement of a stability evaluation the stability protocol should be written and approved—usually by technical services and QA. ● The key elements of a stability protocol include 1. Product name and packaging details. The information should be sufficiently detailed to clearly identify the specific formulation(s) to be evaluated, the specific container/closure types (and sources), the batch size(s). 2- The storage condition. 3. Number of batches to be evaluated. Normally a minimum of three batches is required to provide a sufficient basis for shelf-life prediction. Development and stability batches may be used provided they are of the same formulations as the commercial product and they were processed in an equivalent manner

23
In general, “significant change” for a drug product is defined as:
1. A 5% change in assay from its initial value; or failure to meet the acceptance criteria for potency when using biological or immunological procedures; 2. Any degradation product’s exceeding its acceptance criterion; 3. Failure to meet the acceptance criteria for appearance, physical attributes, and functionality test (e.g., color, phase separation, resuspendibility, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g., softening of suppositories, melting of creams) may be expected under accelerated conditions; and, as appropriate for the dosage form: 4. Failure to meet the acceptance criterion for pH; or 5. Failure to meet the acceptance criteria for dissolution for 12 dosage units
; however, some changes in physical attributes (e.g., softening of suppositories, melting of creams) may be expected under accelerated conditions; and, as appropriate for the dosage form: 4. Failure to meet the acceptance criterion for pH; or. 5. Failure to meet the acceptance criteria for dissolution for 12 dosage units.")
24
ICH used the climatic zone concept
The key points included: • Stability storage conditions will normally involve long-term studies at 25° ± 2°C with 60% RH ± 5% with at least 12 months of data before filing; accelerated studies at 40° ± 2°C and 75% RH ± 5% with at least 6 months of data. • Where ‘‘significant change’’ occurs during the 40°C accelerated study an additional intermediate station should be used, such as 30° ± 2°C/ 60% RH ± 5%. ‘‘Significant change’’ was defined as a 5% loss of potency, any degradant exceeding its specification limit, exceeding pH limits, dissolution failures using 12 units, failures of physical specifications (hardness, color, etc.)
")
25
4- . Test methodology. The stability testing monograph need not include all of the criteria defined in the product release monograph. Only those parameters that are potentially susceptible to change during storage and that may impact on quality, safety, or efficacy need to be evaluated. 5. Test frequency should be adequate to demonstrate any degradation and to provide enough data points for statistical evaluation. For the scale-up batches and the first three commercial batches testing is expected initially, at 3-month intervals during the first year, 6-monthly in the second year, and yearly thereafter

26
• For less stable products the storage (and labeling) conditions may be reduced but the accelerated conditions should still be at least 15°C above those used for long-term evaluation. • For products where water loss may be important, such as liquids or semisolids in plastic containers, it may be more appropriate to replace the high-RH conditions by lower RH such as 10–20%. • The same storage conditions are to be applied for the evaluation of bulk drug substances. However, retest dates may be used instead of expiration dates.

27
For long term studies, frequency of testing should be sufficient to establish the stability profile of the drug product. For products with a proposed shelf life of at least 12 months, the frequency of testing at the long term storage condition should normally be every 3 months over the first year, every 6 months over the second year, and annually thereafter through the proposed shelf life. At the accelerated storage condition, a minimum of three time points, including the initial and final time points (e.g., 0, 3, and 6 months), from a 6-month study is recommended.
, from a 6-month study is recommended.")
28
When testing at the intermediate storage condition is called for as a result of significant
change at the accelerated storage condition, a minimum of four time points, including the initial and final time points (e.g., 0, 6, 9, 12 months), from a 12-month study is recommended.
, from a 12-month study is recommended.")
29
6. Name and/or titles of those responsible for assessing the data
6. Name and/or titles of those responsible for assessing the data. Where possible, and appropriate, the data should be evaluated statistically to obtain the shelf-life

30
Stability studies can be classified into three types: 1
Stability studies can be classified into three types: 1. Studies, usually under accelerated conditions to predict a tentative shelf-life for a new or modified product or process. For a new drug substance these studies usually commence with a preformulation evaluation. The effect of stress conditions such as temperature, humidity, light, acidity, and oxygen, can provide much useful information to the formulator. The potential interactive effects of the bulk drug and the anticipated dosage form excipients may also be evaluated. The accelerated studies at elevated temperature on the dosage form should allow some extrapolation to provide a tentative shelf-life. The ICH guidelines allow extrapolation of 6 months data under accelerated conditions with 12 months data at 25°C/60% RH to predict a shelf-life of up to 24 months. Shelf-life in excess of 24 months should rarely be extrapolated from accelerated data

31
Where there is a change of manufacturing facility for the dosage form, but using the same process and similar equipment, 3 months accelerated data may suffice, again with the commitment to monitor the first three commercial batches

32
2. Studies under conditions appropriate to the market, or those defined in the product labeling, are used to provide real-time data for confirmation of the predicted tentative shelf-life. These studies are usually performed using controlled environmental cabinets. A typical warehouse may be an acceptable alternative provided temperature and humidity are recorded. For certain physical parameters such as dissolution, tablet fragility, and parenteral sterility, accelerated conditions may not provide useful data for extrapolation. Where such studies demonstrate that the predicted tentative shelf-life was too optimistic it would be necessary to consider recall of released batches. Real-time studies are also used to extend the defined shelf-life where the predicted value is found to be too pessimistic.

33
3. Stability studies on current production
3. Stability studies on current production. Once the shelf-life is established it is necessary to evaluate some ongoing batches to confirm that current production is behaving in a similar manner. This is to detect the possible impact of any subtle or unknown changes to the components or process. In the event that a change is observed, it will be necessary to perform a root cause analysis. At this stage there should be a considerable amount of available stability data that identify the shelf-life limiting factors

34
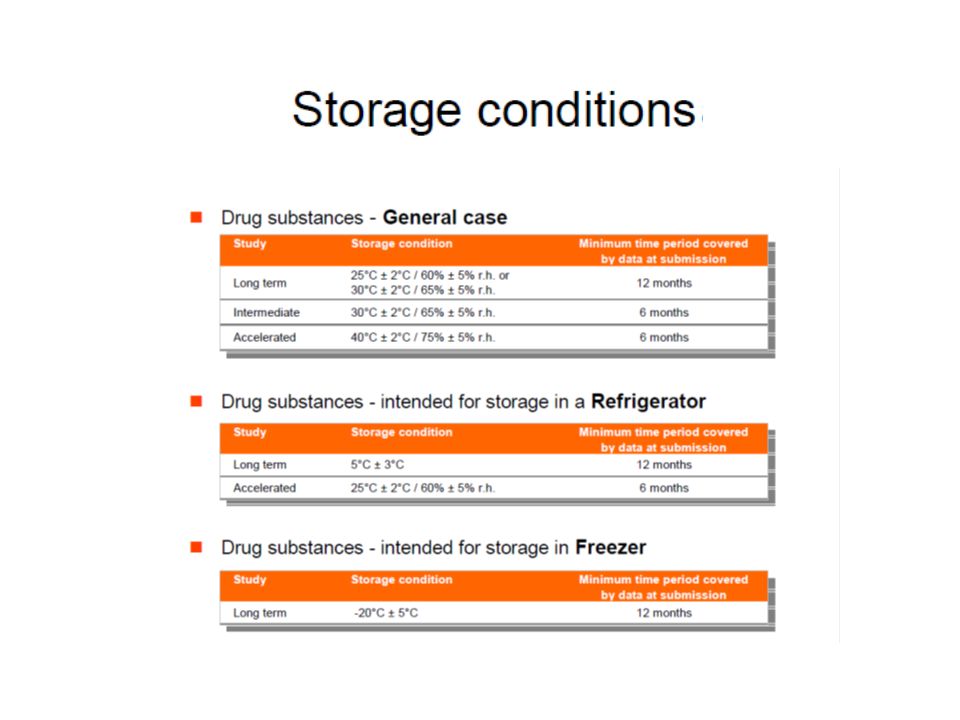
Storage conditions for general products
*It is up to the applicant to decide whether long term stability studies are performed at 25° ± 2°C with 60% RH ± 5% or 30 ° ± 2°C with 65% RH ± 5%. **If 30 ° ± 2°C with 65% RH ± 5% is the long term conditions, there is no intermediate conditions

35
The stability requirements for homeopathic products are less demanding than for other drug products. The levels of ‘‘active ingredients’’ are frequently so low that determination of degradation products, or even assay of the active itself, may not be practicable. The requirements allow examination for compatibility as an alternative to testing.

38
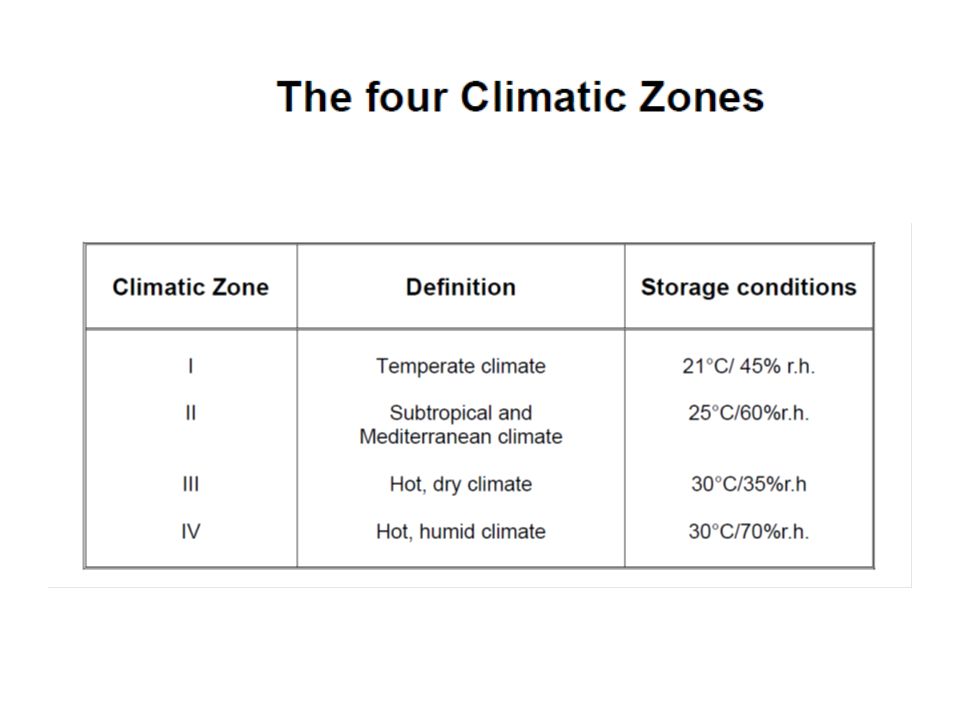
● Climatic zones The four zones in the world that are distinguished by their characteristic prevalent annual climatic conditions.

41
Expiration date The date placed on the container label of a drug product designating the time prior to which a batch of the product is expected to remain within the approved shelf life specification if stored under defined conditions, and after which it must not be used. Shelf life (also referred to as expiration dating period) The time period during which a drug product is expected to remain within the approved shelf-life specification, provided that it is stored under the conditions defined on the container label.
 The time period during which a drug product is expected to remain within the approved shelf-life specification, provided that it is stored under the conditions defined on the container label.")
Similar presentations












. You can freely download, adapt, and distribute this.>")










