Download presentation
Presentation is loading. Please wait.

1
Multiple sequence alignment MSA
Edgar RC, Batzoglou S. Multiple sequence alignment. Curr Opin Struct Biol Jun;16(3): PubMed PMID: Prednaska zpracovana dle Pevsner, Bioinformatics and functional genomics
: PubMed PMID: Prednaska zpracovana dle Pevsner, Bioinformatics and functional genomics.")
2
What is MSA Comparison of many (i.e., >2) sequences local or global
 sequences local or global")
3
Why MSA Biological sequences often occur in families. Homologous sequences often retain similar structures and functions. related genes within an organism genes in various species sequences within a population (polymorphic variants) MSA reveals more biological information than pairwise alignment two sequences that may not align well to each other can be aligned via their relationship to a third sequence, thereby integrating information in a way not possible using only pairwise alignments Similar genes are conserved across widely divergent species, often performing a similar or even identical function, and at other times, mutating or rearranging to perform an altered function through the forces of natural selection. Thus, many genes are represented in highly conserved forms in organisms. Through simultaneous alignment of the sequences of these genes, sequence patterns that have been subject to alteration may be analyzed. For example, it allows the identification of conserved sequence patterns and motifs in the whole sequence family, which are not obvious to detect by comparing only two sequences. Many conserved and functionally critical amino acid residues can be identified in a protein multiple alignment. Multiple sequence alignment is also an essential prerequisite to carrying out phylogenetic analysis of sequence families and prediction of protein secondary and tertiary structures. Homologous residues are aligned in columns across the length of the sequences. These aligned residues are homologous in an evolutionary sense: they are presumably derived from a common ancestor. The residues in each column are also presumed to be homologous in a structural sense: aligned residues tend to occupy corresponding positions in the three-dimensional structure of each aligned protein.
 MSA reveals more biological information than pairwise alignment. two sequences that may not align well to each other can be aligned via their relationship to a third sequence, thereby integrating information in a way not possible using only pairwise alignments. Similar genes are conserved across widely divergent species, often performing a similar or even identical function, and at other times, mutating or rearranging to perform an altered function through the forces of natural selection. Thus, many genes are represented in highly conserved forms in organisms. Through simultaneous alignment of the sequences of these genes, sequence patterns that have been subject to alteration may be analyzed. For example, it allows the identification of conserved sequence patterns and motifs in the whole sequence family, which are not obvious to detect by comparing only two sequences. Many conserved and functionally critical amino acid residues can be identified in a protein multiple alignment. Multiple sequence alignment is also an essential prerequisite to carrying out phylogenetic analysis of sequence families and prediction of protein secondary and tertiary structures. Homologous residues are aligned in columns across the length of the sequences. These aligned residues are homologous in an evolutionary sense: they are presumably derived from a common ancestor. The residues in each column are also presumed to be homologous in a structural sense: aligned residues tend to occupy corresponding positions in the three-dimensional structure of each aligned protein.")
4
Edgar R.S. et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature (7399):459-64
:")
5
LUCA - last universal common ancestor
Edgar R.S. et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature (7399):459-64
:")
6
Why MSA Can be a reasonable way to infer gene function
Characterize protein families by identifying shared regions of homology (conserved regions called motifs), such as active sites Determine the consensus sequence of several aligned sequences Establish relationships and phylogenies
, such as active sites. Determine the consensus sequence of several aligned sequences. Establish relationships and phylogenies.")
7
What is a sequence motif?
A short conserved region in DNA, RNA or protein sequence simple combinations of secondary structure elements motif = supersecondary structure in proteins, structure motifs usually consist of just a few elements; e.g., the 'helix-turn-helix' has just three Corresponds to a structural or functional feature in proteins Shared by several sequences, can be generated by MSA

8
Examples of motifs beta hairpin helix-loop-helix greek key
HLH - Two α-helices (blue) are connected by a shortloop (red) beta hairpin - Two antiparallel beta strands connected by a tight turn of a few amino acids between them greek key - 4 beta strands folded over into a sandwich shape.
 are connected by a shortloop (red) beta hairpin - Two antiparallel beta strands connected by a tight turn of a few amino acids between them. greek key - 4 beta strands folded over into a sandwich shape.")
9
What is a protein family?
A protein family is a group of evolutionarily-related proteins. Proteins in a family descend from a common ancestor and typically have similar three-dimensional structures, functions, and significant sequence similarity. Members of a protein family may range from very similar to quite diverse. Currently, over 60,000 protein families have been defined, although ambiguity in the definition of protein family leads different researchers to wildly varying numbers

10
What is a protein family?
the use of protein family is somewhat context dependent A common usage is that superfamilies (structural homology) contain families (sequence homology) which contain sub-families. Example: superfamily PA clan the largest group of proteases with common ancestry as identified by structural homology has far lower sequence conservation than one of the families tit contains, the C04 family
 contain families (sequence homology) which contain sub-families. Example: superfamily PA clan. the largest group of proteases with common ancestry as identified by structural homology. has far lower sequence conservation than one of the families tit contains, the C04 family.")
12
PA clan structure the double-beta barrel motif
PA clan proteases all share a core motif of two β-barrels with covalent catalysis performed by an acid-histidine-nucleophile catalytic triad motif. Structural homology in the PA superfamily. The double beta-barrel that characterises the superfamily is highlighted in red. Shown ate representative structures from several families within the PA superfamily. Note that some proteins show partially modified structural. Chymotrypsin (1gg6), thrombin(1mkx), tobacco etch virus protease (1lvm), calicivirin (1wqs), west nile virus protease (1fp7), exfoliatin toxin (1exf), HtrA protease (1l1j), snake venom plasminogen activator (1bqy), chloroplast protease (4fln) and equine arteritis virus protease (1mbm).
, thrombin(1mkx), tobacco etch virus protease (1lvm), calicivirin (1wqs), west nile virus protease (1fp7), exfoliatin toxin (1exf), HtrA protease (1l1j), snake venom plasminogen activator (1bqy), chloroplast protease (4fln) and equine arteritis virus protease (1mbm).")
13
Pfam - http://pfam.sanger.ac.uk/
Database of protein families that includes their annotations and multiple sequence alignments

14
What is a domain Families often share domains. Domain is a part of a protein, and is greater than a motif. Domain is formed by several motifs packed together. i.e. domain = tertiary structure Domain - a conserved part of a given protein sequence and structure that can evolve, function, and exist independently of the rest of the protein chain One domain may appear in a variety of different proteins Molecular evolution uses domains as building blocks and these may be recombined in different arrangements to create proteins with different functions

15
Pyruvate kinase domains
Because they are independently stable, domains can be "swapped" by genetic engineering between one protein and another to make chimeric proteins.

16
Sequence logo Conserved: BIG letters with few others in that space
Divergent: small letters with many others in that space

17
Logo and alignment reflect each other

18
Doing MSA As with aligning a pair of sequences, the difficulty in aligning a group of sequences varies considerably with sequence similarity. If the amount of sequence variation is minimal, it is quite straightforward to align the sequences, even without the assistance of a computer program. If the amount of sequence variation is great, it may be very difficult to find an optimal alignment of the sequences because so many combinations of substitutions, insertions, and deletions, each predicting a different alignment, are possible.

19
Challenges of the MSA Finding an optimal alignment of more than two sequences that includes matches, mismatches, and gaps, and that takes into account the degree of variation in all of the sequences at the same time poses a very difficult challenge. A second computational challenge is identifying a reasonable method of obtaining a cumulative score for the substitutions in the column of an MSA. Finally, the placement and scoring of gaps in the various sequences of an msa presents an additional challenge.

20
MSA algorithms As with the pairwise sequence comparisons, there are two types of multiple alignment algorithms optimal heuristic

21
Optimal algorithms Extension of dynamic programming to multiple sequences Exhaustive search Produce best alignment Computationally expensive Not feasible for n>10 sequences of length m>200 residues

22
Heuristic algorithms Limit the exhaustive search
Attempt to rapidly find a good, but not necessarily optimal alignment Most popular methods: progressive methods (ClustalW) start from the most similar sequences and progressively add new sequences iterative methods (MUSCLE) make initial crude alignment, then revise it
 start from the most similar sequences and progressively add new sequences. iterative methods (MUSCLE) make initial crude alignment, then revise it.")
23
Progressive sequence alignment
The most commonly used algorithm, the most commonly used software ClustalW Popularized by Feng and Doolitle, often referred by these two names. Permits the rapid alignment of even hundreds of sequences. Limitation: the final alignment depends on the order in which sequences are joined. Not guaranteed to provide the most accurate alignments.

24
ClustalW http://www.clustal.org
EMBOSS – a free open source software analysis package (European Molecular Biology Open Software Suite) - Program emma is a ClustalW wrapper A variety of EMBOSS servers hosting emma are available, e.g. ClustalX – a downloadable stand-alone program offering a graphical user interface for editing multiple sequence alignments - ClustalW paper:
 - Program emma is a ClustalW wrapper. A variety of EMBOSS servers hosting emma are available, e.g. ClustalX – a downloadable stand-alone program offering a graphical user interface for editing multiple sequence alignments. - ClustalW paper:")
27
ClustalW – how it works? Three stages

28
number of pairwise alignments
1st stage The global alignment (Needlman-Wunsch) is used to create pairwise alignments of every protein pair. number of pairwise alignments 𝑛(𝑛−1) 2 arrow shows the best score
 is used to create pairwise alignments of every protein pair. number of pairwise alignments. 𝑛(𝑛−1) 2. arrow shows the best score.")
29
1st stage The raw similarity scores are shown. However, for the next step the distance matrix is needed, and not the similarity one. Similarity scores must be converted into distances. Won't tell you how, believe me, it is doable.

30
2nd stage A guide tree is calculated from the distance matrix.
The tree reflects the relatedness of all the proteins to be multiply aligned Newick format

31
Guided tree Guide trees are not true phylogenetic trees.
They are templates used in the third stage of ClustalW to define the order in which sequences are added to a multiple alignment. A guided tree is estimated from a distance matrix of the sequences you are aligning. In contrast, a phylogenetic tree almost always includes a model to account for multiple substitutions that commonly occur at the position of aligned residues.

32
Construction of guided tree
Unweighted Pair Group Method with Arithmetic Mean (UPGMA) A simple hierarchical clustering method How it works? Neighbor joining Uses distance method, distance matrix is an input. The algorithm starts with a completely unresolved tree (its topology is a star network), and iterates over until the tree is completely resolved and all branch lengths are known. Dr. Richard Ewdards - UPGMA Worked Example UPGMA = hierarchical clustering, average linkage
 A simple hierarchical clustering method. How it works Neighbor joining. Uses distance method, distance matrix is an input. The algorithm starts with a completely unresolved tree (its topology is a star network), and iterates over until the tree is completely resolved and all branch lengths are known. Dr. Richard Ewdards - UPGMA Worked Example. UPGMA = hierarchical clustering, average linkage.")
33
From http://en.wikipedia.org/wiki/Neighbor_joining:
Starting with a star tree (A), the Q matrix is calculated and used to choose a pair of nodes for joining, in this case f and g. These are joined to a newly created node, u, as shown in (B). The part of the tree shown as dotted lines is now fixed and will not be changed in subsequent joining steps. The distances from node u to the nodes a-e are computed from the formula given in the text. This process is then repeated, using a matrix of just the distances between the nodes, a,b,c,d,e, and u, and a Q matrix derived from it. In this case u and e are joined to the newly created v, as shown in (C). Two more iterations lead first to (D), and then to (E), at which point the algorithm is done, as the tree is fully resolved. Source: wikipedia
, the Q matrix is calculated and used to choose a pair of nodes for joining, in this case f and g. These are joined to a newly created node, u, as shown in (B). The part of the tree shown as dotted lines is now fixed and will not be changed in subsequent joining steps. The distances from node u to the nodes a-e are computed from the formula given in the text. This process is then repeated, using a matrix of just the distances between the nodes, a,b,c,d,e, and u, and a Q matrix derived from it. In this case u and e are joined to the newly created v, as shown in (C). Two more iterations lead first to (D), and then to (E), at which point the algorithm is done, as the tree is fully resolved. Source: wikipedia.")
34
3rd stage The multiple sequence alignment is created in a series of steps based on the order presented in the guide tree. First select the two most closely related sequences from the guide tree and create a pairwise alignment.

35
3rd stage

36
3rd stage The next sequence is either added to the pairwise alignment (to generate an aligned group of three sequences, sometimes called a profile) or used in another pairwise alignment. At some point, profiles are aligned with profiles. The alignment continues progressively until the root of the tree is reached, and all sequences have been aligned.
 or used in another pairwise alignment. At some point, profiles are aligned with profiles. The alignment continues progressively until the root of the tree is reached, and all sequences have been aligned.")
37
Gaps “once a gap, always a gap” rule
The most closely related pair of sequences is aligned first. As further sequences are added to the alignment, there are many ways that gaps could be included. Gaps are often added to first two (closest) sequences. To change the initial gap choices later on would be to give more weight to distantly related sequences. To maintain the initial gap choices is to trust that those gaps are most believable.
 sequences. To change the initial gap choices later on would be to give more weight to distantly related sequences. To maintain the initial gap choices is to trust that those gaps are most believable.")
38
Iterative approaches Progressive alignment methods have the inherent limitation that once an error occurs in the alignment process it cannot be corrected, and iterative approaches can overcome this limitation. Create an initial alignment and then modify it to try to improve it. e.g. MUSCLE, IterAlign, Praline, MAFFT

39
MUSCLE Since its introduction in 2004, the MUSCLE program of Robert Edgar has become popular because of its accuracy and its exceptional speed, especially for multiple sequence alignments involving large number of sequences. Multiple sequence comparison by log expectation Three stages

40
MUSCLE 1. Draft alignment 2. Improved alignment 3. Refinement
Edgar, R. C. Nucl. Acids Res : ; doi: /nar/gkh340

41
1st stage A draft progressive alignment is generated.
Determine pairwise similarity through k-mer counting (not by alignment). Compute distance (triangular distance) matrix. Construct tree using UPGMA. Construct draft progressive alignment following tree.
. Compute distance (triangular distance) matrix. Construct tree using UPGMA. Construct draft progressive alignment following tree.")
42
2nd stage Improve the progressive alignment.
Compute pairwise identity through current MSA using the fractional identity. Construct new tree using Kimura distance matrix. In a comparison of two sequences there is some likelihood that multiple amino acid (or nucleotide) substiutions occurred at any given position, and the Kimura distance matrix provides a model for such changes. Compare new and old trees: if improved, repeat this step, if not improved, then we’re done.
 substiutions occurred at any given position, and the Kimura distance matrix provides a model for such changes. Compare new and old trees: if improved, repeat this step, if not improved, then we’re done.")
43
3rd stage Refinement of the MSA
Systematically partition the tree to obtain subsets; an edge (branch) of the tree is deleted to create a bipartition. Extract a pair of profiles (multiple sequence alignments), and realign them Accept/reject the new alignment based on the sum-of-pairs score increase/decrease. All edges of the tree are systematically visited and deleted to create bipartitions. This iterative refinement step is rapid and had been shown earlier to increase the accuracy of the multiple sequence alignment.
 of the tree is deleted to create a bipartition. Extract a pair of profiles (multiple sequence alignments), and realign them. Accept/reject the new alignment based on the sum-of-pairs score increase/decrease. All edges of the tree are systematically visited and deleted to create bipartitions. This iterative refinement step is rapid and had been shown earlier to increase the accuracy of the multiple sequence alignment.")
44
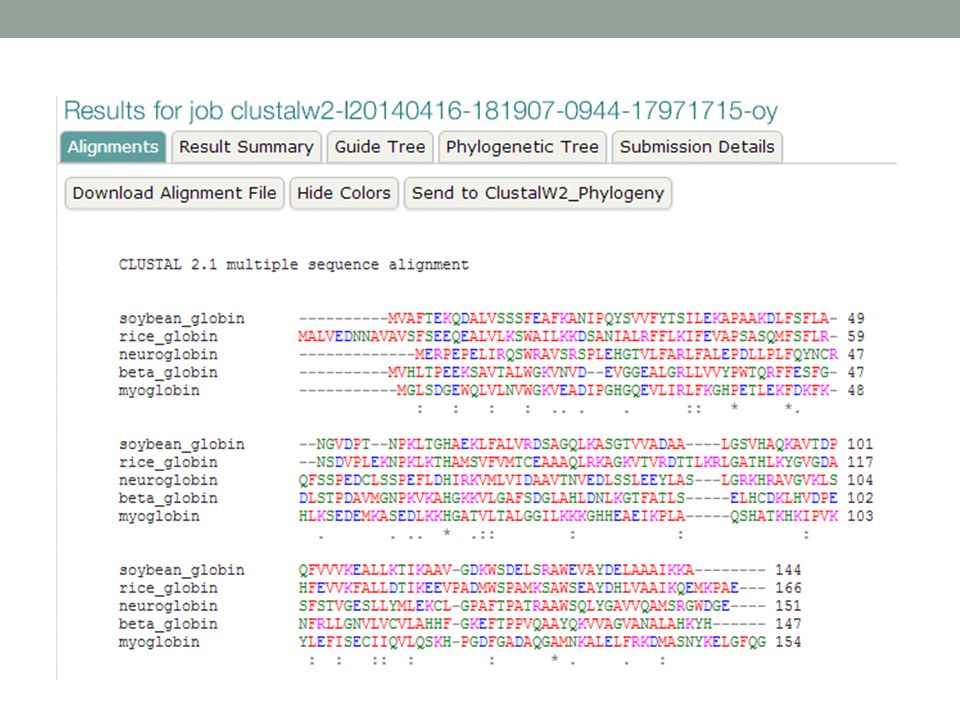
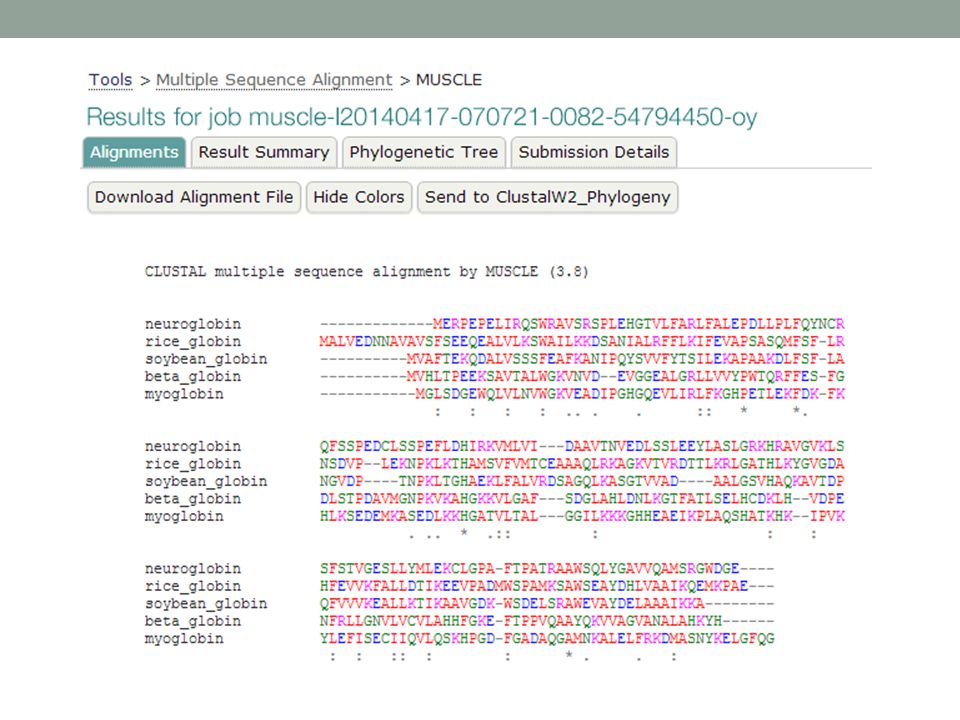
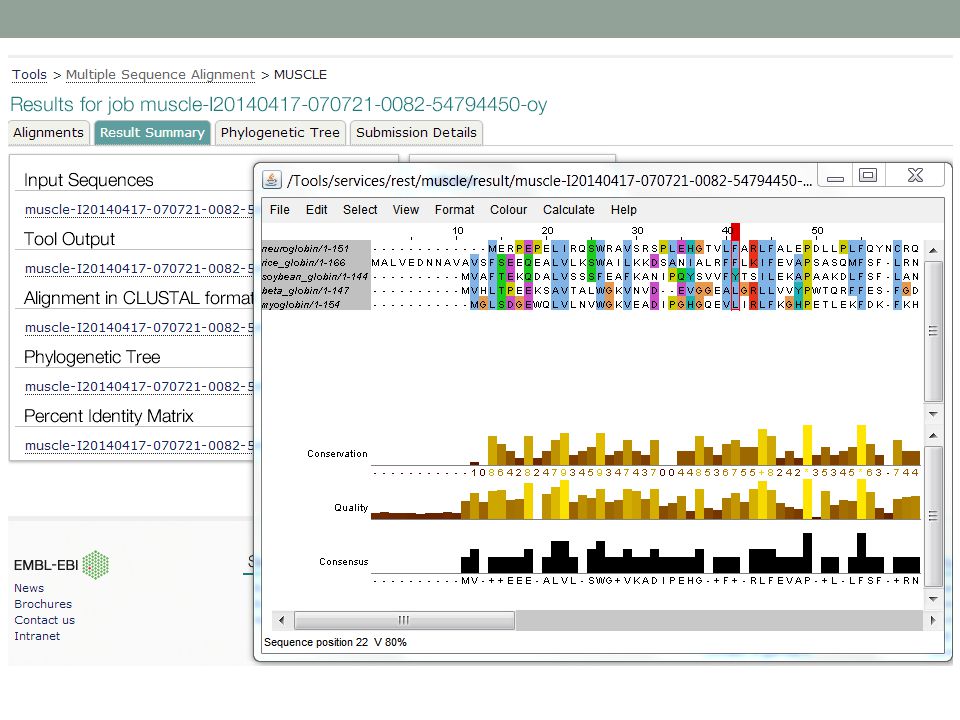
MUSCLE online

47
>neuroglobin 1OJ6A NP_067080
>neuroglobin 1OJ6A NP_ [Homo sapiens] MERPEPELIRQSWRAVSRSPLEHGTVLFARLFALEPDLLPLFQYNCR QFSSPEDCLSSPEFLDHIRKVMLVI---DAAVTNVEDLSSLEEYLASLGRKHRAVGVKLS SFSTVGESLLYMLEKCLGPA-FTPATRAAWSQLYGAVVQAMSRGWDGE---- >rice_globin 1D8U rice Non-Symbiotic Plant Hemoglobin NP_ [Oryza sativa (japonica cultivar-group)] MALVEDNNAVAVSFSEEQEALVLKSWAILKKDSANIALRFFLKIFEVAPSASQMFSF-LR NSDVP--LEKNPKLKTHAMSVFVMTCEAAAQLRKAGKVTVRDTTLKRLGATHLKYGVGDA HFEVVKFALLDTIKEEVPADMWSPAMKSAWSEAYDHLVAAIKQEMKPAE--- >soybean_globin 1FSL leghemoglobin P02238 LGBA_SOYBN [Glycine max] MVAFTEKQDALVSSSFEAFKANIPQYSVVFYTSILEKAPAAKDLFSF-LA NGVDP----TNPKLTGHAEKLFALVRDSAGQLKASGTVVAD----AALGSVHAQKAVTDP QFVVVKEALLKTIKAAVGDK-WSDELSRAWEVAYDELAAAIKKA >beta_globin 2hhbB NP_ [Homo sapiens] MVHLTPEEKSAVTALWGKVNVD--EVGGEALGRLLVVYPWTQRFFES-FG DLSTPDAVMGNPKVKAHGKKVLGAF---SDGLAHLDNLKGTFATLSELHCDKLH--VDPE NFRLLGNVLVCVLAHHFGKE-FTPPVQAAYQKVVAGVANALAHKYH >myoglobin 2MM1 NP_ [Homo sapiens] MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDK-FK HLKSEDEMKASEDLKKHGATVLTAL---GGILKKKGHHEAEIKPLAQSHATKHK--IPVK YLEFISECIIQVLQSKHPGD-FGADAQGAMNKALELFRKDMASNYKELGFQG
] MALVEDNNAVAVSFSEEQEALVLKSWAILKKDSANIALRFFLKIFEVAPSASQMFSF-LR NSDVP--LEKNPKLKTHAMSVFVMTCEAAAQLRKAGKVTVRDTTLKRLGATHLKYGVGDA HFEVVKFALLDTIKEEVPADMWSPAMKSAWSEAYDHLVAAIKQEMKPAE--- >soybean_globin 1FSL leghemoglobin P02238 LGBA_SOYBN [Glycine max] MVAFTEKQDALVSSSFEAFKANIPQYSVVFYTSILEKAPAAKDLFSF-LA NGVDP----TNPKLTGHAEKLFALVRDSAGQLKASGTVVAD----AALGSVHAQKAVTDP QFVVVKEALLKTIKAAVGDK-WSDELSRAWEVAYDELAAAIKKA >beta_globin 2hhbB NP_ [Homo sapiens] MVHLTPEEKSAVTALWGKVNVD--EVGGEALGRLLVVYPWTQRFFES-FG DLSTPDAVMGNPKVKAHGKKVLGAF---SDGLAHLDNLKGTFATLSELHCDKLH--VDPE NFRLLGNVLVCVLAHHFGKE-FTPPVQAAYQKVVAGVANALAHKYH >myoglobin 2MM1 NP_ [Homo sapiens] MGLSDGEWQLVLNVWGKVEADIPGHGQEVLIRLFKGHPETLEKFDK-FK HLKSEDEMKASEDLKKHGATVLTAL---GGILKKKGHHEAEIKPLAQSHATKHK--IPVK YLEFISECIIQVLQSKHPGD-FGADAQGAMNKALELFRKDMASNYKELGFQG.")
48
MUSCLE vs. Clustal Q = fraction of correctly aligned residues (pairwise) TC = fraction of correctly aligned columns
 TC = fraction of correctly aligned columns.")
49
Logo visualization of the alignment
Make a logo from your alignment Can be easier to compare Nice graphic Students love ‘em

Similar presentations














 - search for remote homologs using HMMs or profiles.>")




 The Mechanics of Alignments.>")


