Download presentation
Presentation is loading. Please wait.

1
Quantum Calculations B. Barbiellini bba@neu.edu Thematics seminar April 21,2005

2
Goal: Solve the Schrödinger equation Application: Description of chemical bonds

3
Outline Independent Particle Approximation (IPM) and Hartree Fock (HF) SCF: Basis sets. Other theoretical methods: DFT and QMC. Illustrative example: Study of Hydrogen bond in ice and water.

5
Electronic structure theory H = E Ab-initio - from the origins (First-principles) No experimental parameters Few physical constants c, h, m e, q e
 No experimental parameters Few physical constants c, h, m e, q e")
6
min H| = E Variational Theorem

7
Theoretical Methods SCF & post-SCF methods (CI) Density functional theory (DFT) Stochastic methods: Quantum Monte Carlo (QMC)
 Density functional theory (DFT) Stochastic methods: Quantum Monte Carlo (QMC)")
8
Climbing Mt. Psi Correlation energy: energy contributions beyond SCF

9
= det( r))det( r Independent Particle Model: Hartree-Fock (HF) SCF is a molecular orbital is spin up F =e F is an effective one-particle hamiltonian which depend on MO’s Self Consistent Field (SCF).
)det( r Independent Particle Model: Hartree-Fock (HF) SCF is a molecular orbital is spin up F =e F is an effective one-particle hamiltonian which depend on MO’s Self Consistent Field (SCF).")
10
Linear combination of atomic orbitals termed “basis functions” Basis set – mathematical representation of molecular orbitals Minimal basis set – one basis function for every atomic orbital that is required to describe the free atom H(1s) C(1s,2s,2p) → CH 4 :9 basis functions Larger basis sets are more flexible –better approximation of exact MOs Polarization functions, diffuse functions
 C(1s,2s,2p) → CH 4 :9 basis functions Larger basis sets are more flexible –better approximation of exact MOs Polarization functions, diffuse functions")
11
Slater-type orbitals (J.C. Slater) –Represent electron density well in valence region and beyond (not so well near nucleus) –Evaluating these integrals is difficult Gaussian-type orbitals (F. Boys) –Easier to evaluate integrals, but do not represent electron density well –Overcome this by using linear combination of GTOs STOs v. GTOs
 –Represent electron density well in valence region and beyond (not so well near nucleus) –Evaluating these integrals is difficult Gaussian-type orbitals (F. Boys) –Easier to evaluate integrals, but do not represent electron density well –Overcome this by using linear combination of GTOs STOs v. GTOs.")
12
Density functional theory Less expensive than post-SCF methods Include some electron correlation E elec = E T + E V + E J + E XC Pure functionals: BP86, BLYP Hybrid HF/DFT: B3LYP Good for geometries, electron affinities Good for large systems Problem: not systematic

13
Example:Gaussian Input #RHF/6-31G(d) Pop=Full Test RHF/6-31G(d) formaldehyde single point 0,1 C 0.0 0.0 0.0 O 0.0 1.22 0.0 H 0.94 -0.54 0.0 H -0.94 -0.54 0.0 method basis set key words } route section blank line } title section charge, multiplicity } molecular structure section atomic symbols (or numbers) xyz coordinates (or z-matrix) blank line
 Pop=Full Test RHF/6-31G(d) formaldehyde single point 0,1 C O H H method basis set key words } route section blank line } title section charge, multiplicity } molecular structure section atomic symbols (or numbers) xyz coordinates (or z-matrix) blank line")
14
Quantum Monte Carlo Deals with the many body wave-function. Include electron correlation (Jastrow terms). Variation QMC --- Stochastic Gradient Approximation (SGA). Diffusion QMC (almost exact, fixed node approximation) --- computational expensive.
. Variation QMC --- Stochastic Gradient Approximation (SGA). Diffusion QMC (almost exact, fixed node approximation) --- computational expensive..")
18
Distance H-H

22
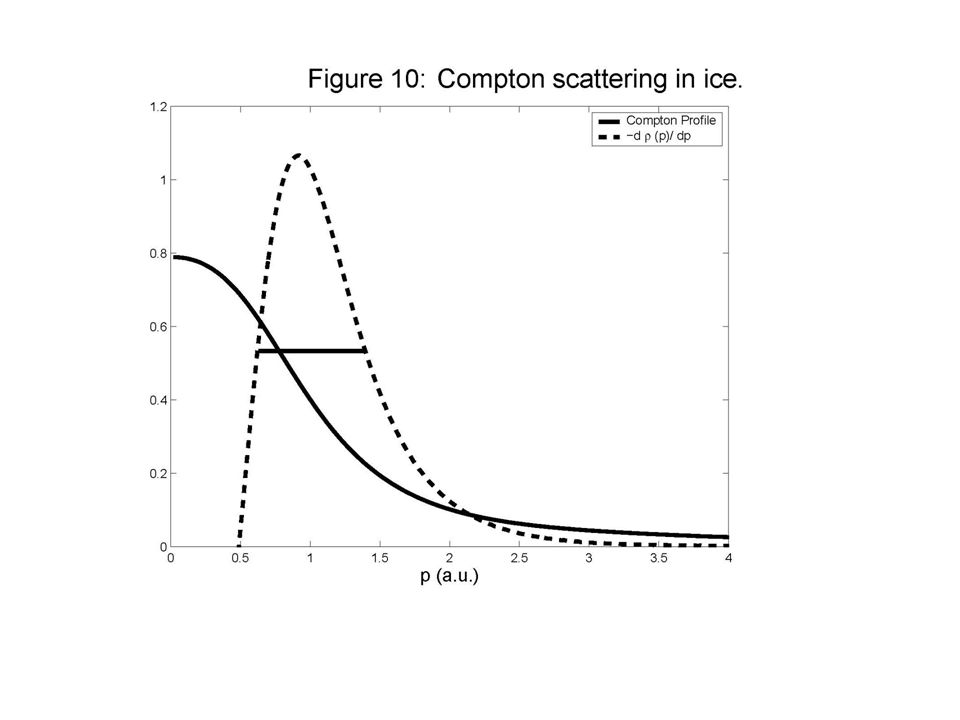
Scattered x rays in ice Isaacs et al., PRL 82 (1999) 600
 600")
23
Wavelike fringes corresponding to interference between the electrons on neighboring sigma and hydrogen bonding sites Compton Profile Anisotropy

26
B(r) Fourier transform CP: MO orbital autocorrelation function
 Fourier transform CP: MO orbital autocorrelation function")
27
Conclusion Quantum calculations are of interest because they can deal with electronic effects, electron de-localization, charge-transfer, and other phenomena, which are otherwise difficult or impossible to treat at the level of classical mechanics.

Similar presentations









 Workshop.>")











! Hψ = Eψ A differential (operator) eigenvalue equation H.>")


 Lewis structures and octet rule>")


 CCSDT … Full CI Minimal Split-valence Polarized Diffuse.>")
 www2.le.ac.uk/departments/physics/people/mervynroy.>")